-
 TechBlog
TechBlog
BasepairでのDESeq2パイプライン
DESeq2パイプラインが実行されると、最初にExpression count(STAR)パイプラインが自動的に実行されます。DESeq2パイプラインはSTARパイプラインで生成された遺伝子カウントマトリックスを使用しま […] -
TechBlog
Reactomeパスウェイエンリッチメント解析のアウトプットファイル
「DESeq2」パイプラインによって生成された全てのアウトプットファイルは「Input/output」タブ(赤枠 1)にあります。Reactome のパスウェイエンリッチメント結果は、「pathway […] -
TechBlog
Reactomeパスウェイエンリッチメント解析からアウトプットされたGSEAレポート
DESeq2パイプラインでパスウェイエンリッチメント解析を実行するとGSEAレポートが作成されます。このレポートはzipフォルダ(図1の青枠)にあります。このフォルダは「Input/output」タブ(図1の赤枠)からア […] -
TechBlog
Basepairでパスウェイ解析(Reactome)
パスウェイ解析とは Basepairではパスウェイ解析も行うことができます。あらかじめ定義された遺伝子や遺伝子産物の位置、遺伝子同士の相互作用などの情報を含んだモデルをもとに行うため、より生物学的な議論を行う […] -
TechBlog
BasepairでのSingle cell RNA-seq integrateパイプラインのアウトプットファイル
IntegrateパイプラインでSeuratが生成するアウトプットファイルについて説明します。アウトプットファイルは、「Input/output」タブ(下図の赤枠)にあります。 Seurat ファイル名ファイルの説明se […] -
TechBlog
BasepairでのSingle cell RNA-seq integrateパイプライン
Integrateパイプラインは、異なるシングルセルRNA-seqサンプルを比較するために使用します。ここでは、シングルセルRNA-seqパイプラインで生成された各サンプルのSeurat Objectをインプットとして使 […] -
TechBlog
BasepairでのSingle cell RNA-seqパイプラインのアウトプットファイル
パイプラインのアウトプットファイルは、「Input/output」タブにあります。 Alevin ファイル名ファイルの説明extract/out1/alevin/whitelist.txtデータから特定された有効な細胞の […] -
TechBlog
BasepairでのSingle cell RNA-seqパイプライン
シングルセルRNA-seqパイプラインではfastpを用いてQCとトリミングを行います。まず、UMI-toolsを用いてバーコードとユニーク分子識別子(UMI)を抽出します。次に、STARを用いてアライメントを行います。 […] -
 TechBlog
TechBlog
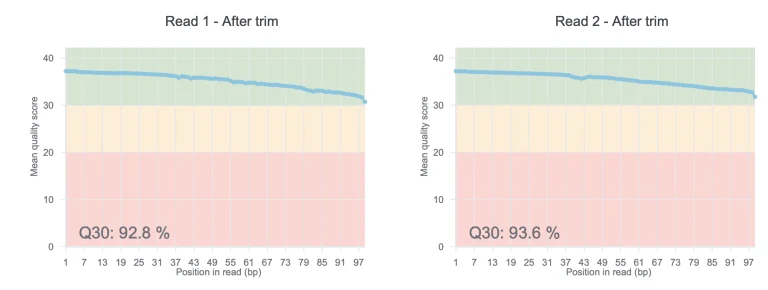
FASTQファイルの品質評価と前処理
なぜFASTQの品質が重要か FASTQの品質は、下流の解析の品質に影響します。 FASTQデータには、低品質リードが含まれることがあります。多くの場合、これはシーケンシングエラーやPCRバイアスなどの要因によって生じま […] -
TechBlog
Basepairでのエピジェネティクスデータのモチーフ検出とアノテーション
はじめに モチーフ検出とは、繰り返し現れる短いDNA配列を検出することです。エピジェネティクスでは、転写因子や他の制御タンパク質の結合部位を表します。 モチーフの検出はピークコーリングの後に行われます(図1)。ピークコー […]
