
BasepairのシングルセルRNA-Seq
fastqがあれば、Basepairを使うことで、シングルセルRNA-Seqの解析を迅速に行うことができます。一気にデータQCやアライメントから視覚化まで完了します。
fastq(.gz)をアップロードするだけで完了
BasepairのシングルセルRNA-Seqでは、fastq(.gz)をアップロードするだけで、自動的に以下のワークフローを実行します。ファイルフォーマットを気にしたり、パイプライン同士を繋げたりする必要はありません。
- データQC
- トリミング
- アライメント
- リードカウント
- 次元削減・クラスタリング(PCA・t-SNE・UMAP)
査読済みツールで構築
すべてのパイプラインは、引用度の高い、査読済みのツールを使用して構築されています。
- fastp:QC
- STAR:アライメント
- UMI tools:バーコードを抽出
- Seurat v3:クラスタリングと視覚化
また、Basepairには、ユーザーが処理群と対照群(コントロール)の両方に複数の複製をアップロードできる「Single cell RNA-seqintegrate」と呼ばれるパイプラインがあります。 Basepair はすべてのサンプルを個別に分析し、これらのサンプルをカウントマトリックスに組み合わせて、2つのグループ間で発現の異なるクラスターを表示します。

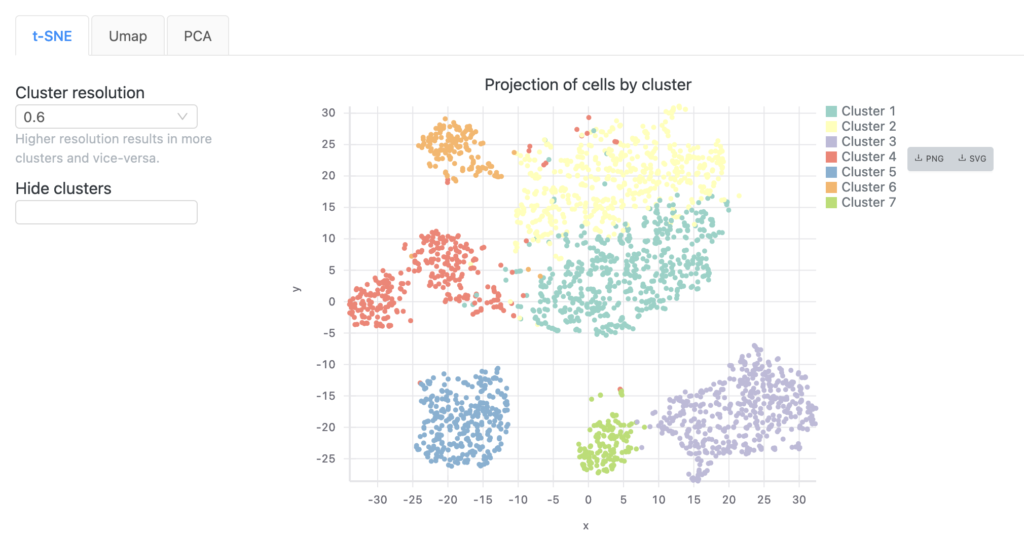
クラスタリングと視覚化
インタラクティブなシングルセルRNA-seq 解析レポートには、t-SNE、UMAP、および PCA プロットによるクラスタリングと視覚化が含まれています。
t-SNE、UMAP、PCAはタブによって切り替えることができ、クラスターの解像度をプルダウンで選択することができます。
プロットはPNG、SVGでダウンロードすることができます。
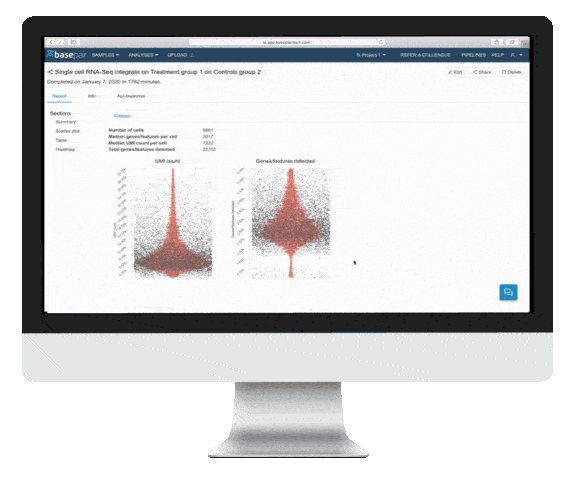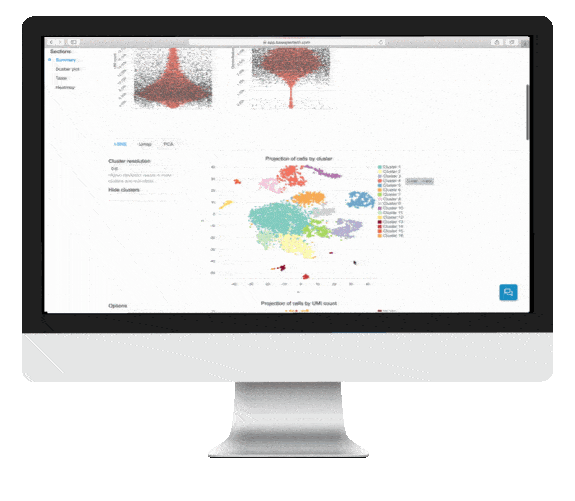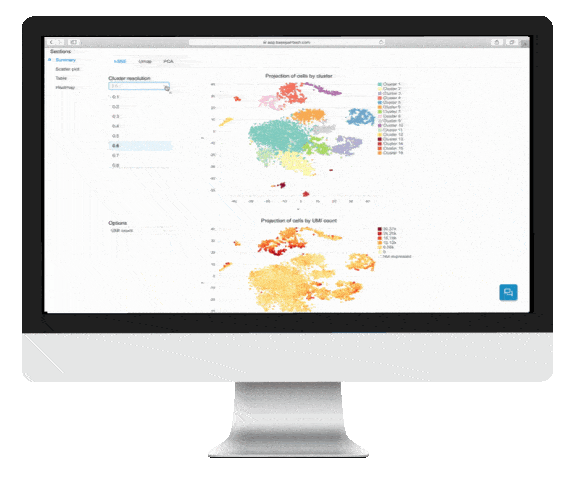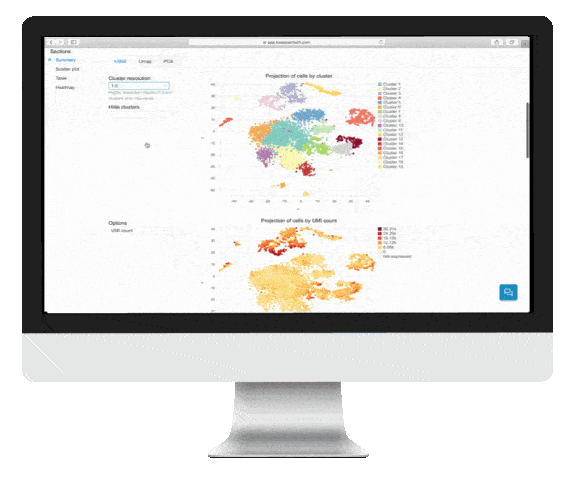
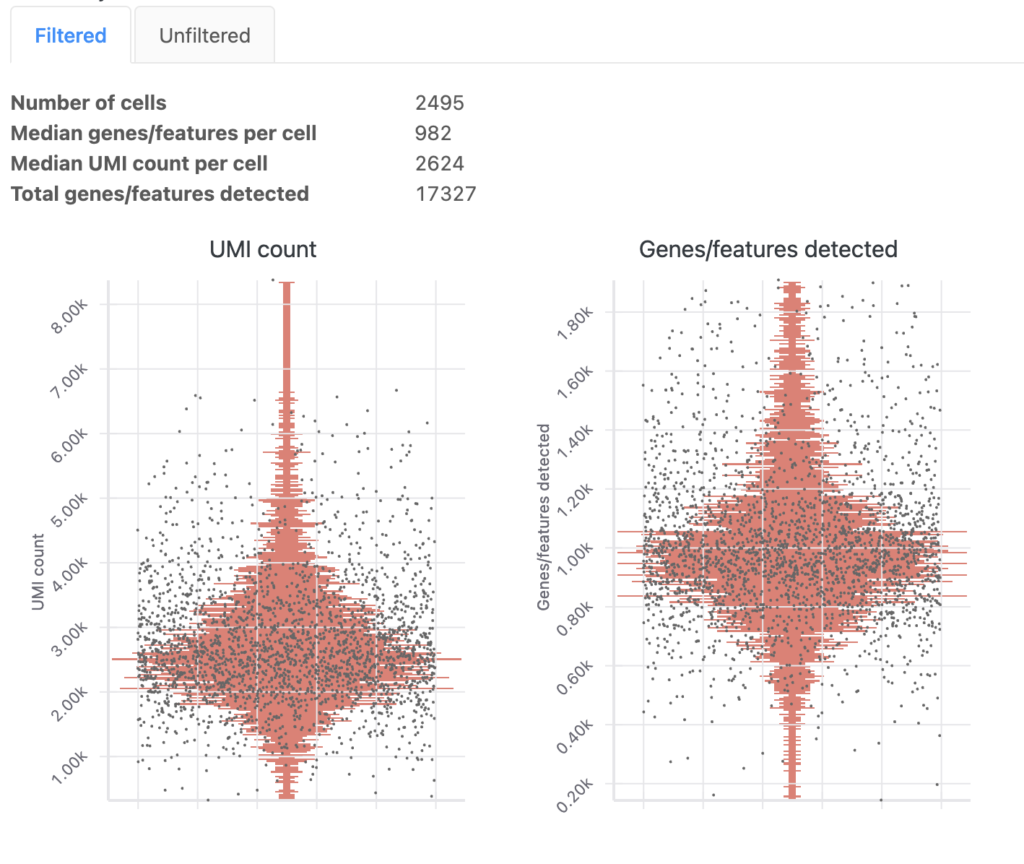
データQC
Basepair のシングルセルRNA-seq 解析レポートには、細胞ごとの品質メトリクスを示すプロットが含まれています。
- 検出された遺伝子/特徴は、 細胞ごとに検出された固有の遺伝子の数を示します。 固有の遺伝子が非常に低い場合は空の液滴を示す可能性があり、非常に高い値は 2 つ以上の細胞を含む液滴を示す場合があります。
- UMI カウントは、 細胞ごとに検出された固有の分子の数を示します。 非常に低い値または高い値は、固有の遺伝子の結果と同様の結果を示します。
- ミトコンドリア比率は 、ミトコンドリア ゲノムにマッピングされているリードの比率です。 0.1 より大きい値は、低品質のセルまたは死にかけているセルを示している可能性があります。
一般に、外れ値を削除し、セルが1つのグループに集まっていることを確認したいと考えます。 適切なデフォルトのフィルタリングしきい値が提供されていますが、結果を自分で評価できるように、フィルタリングの前後のデータも表示されます。
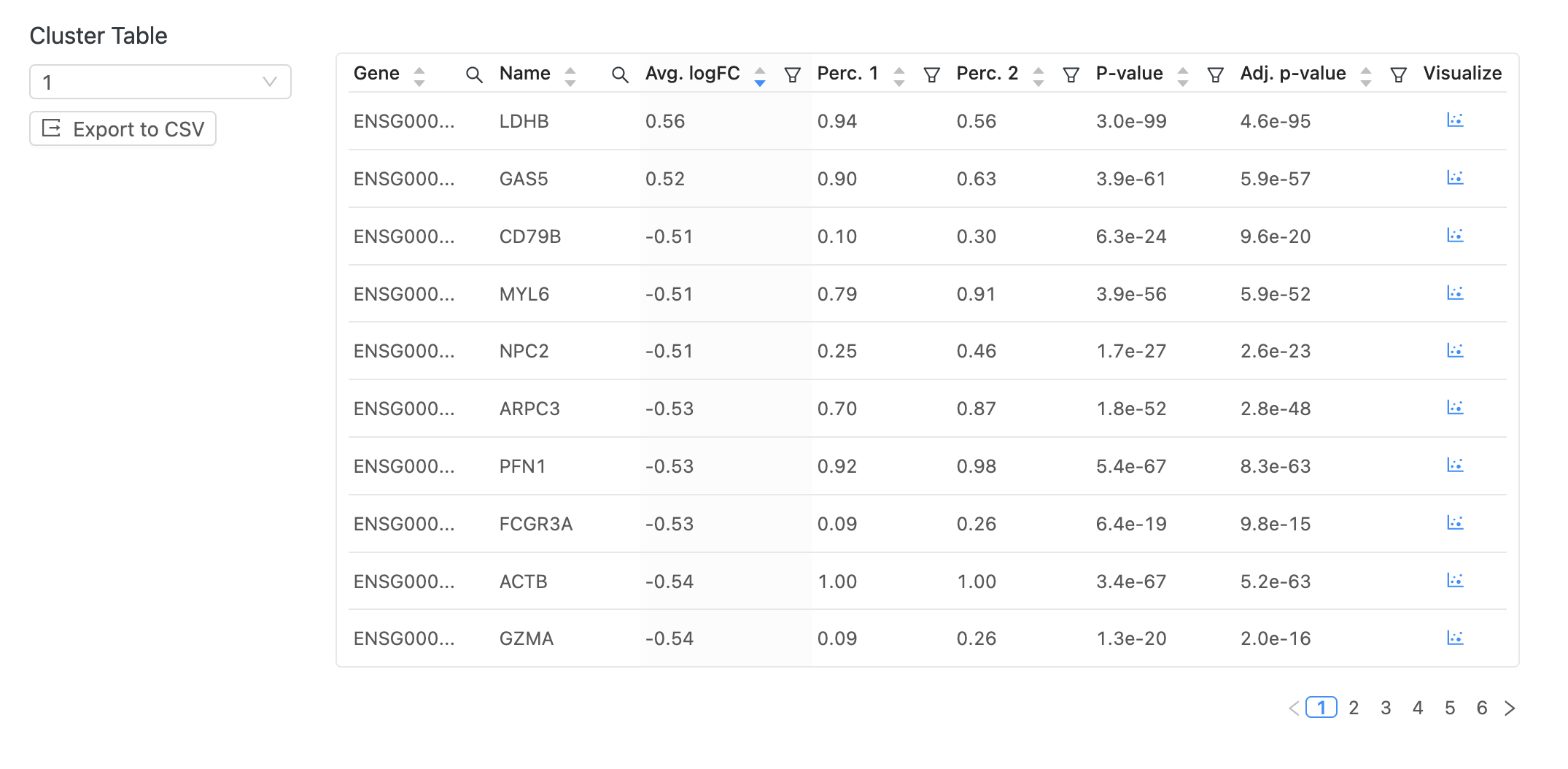
発現差異解析(Differential Expression)
発現差異解析は、t-SNE および UMAP プロットの各クラスターで独自にアップレギュレーとまたはダウンレギュレーとされる遺伝子を示します。 分析では、細胞の各クラスターを他のすべてのクラスターと比較し、各遺伝子の log2 倍率変化、p値、および調整されたp値を出力します。 ここで特に役立つ指標は、log2倍数変化と調整されたp値です。 これらはそれぞれ、遺伝子発現の変化の大きさを示し、偽陽性の可能性を減らします。


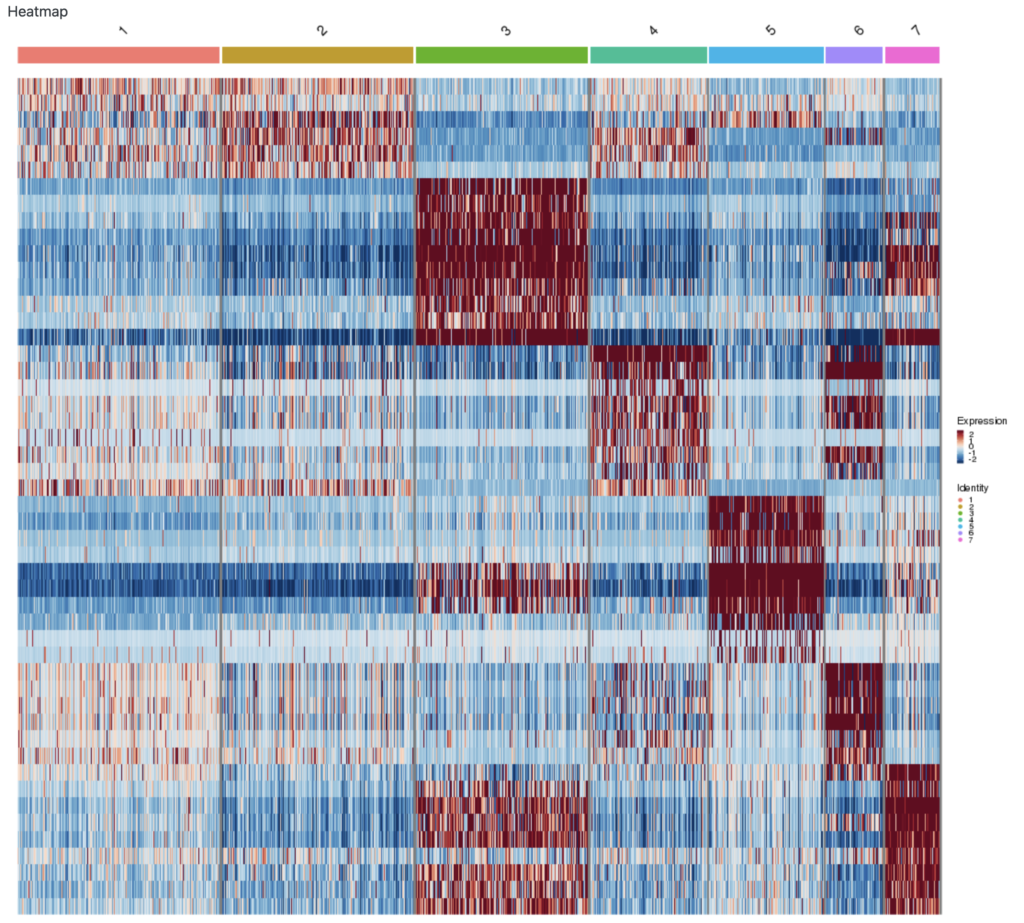
ヒートマップ
t-SNE および UMAP プロットのクラスターごとに最もアップレギュレーとされた遺伝子の上位10個を視覚化するヒートマップが含まれています。 その主な目的は、クラスターを分離するために選択された遺伝子の識別力を視覚化することです。 ヒートマップの追加の便利な機能は、互いに区別するのは難しいものの、同じ種類のセルに対応する可能性のあるクラスターを強調表示できることです。
よくある質問
シングルセルRNA-seq データは、単一細胞分析パイプラインを使用して Basepair 上で迅速に分析できます。 アカウントを作成し 、fastq ファイルをアップロードして、[実行] をクリックします。 パイプラインは自動的に読み取りを調整し、バーコードを抽出し、データをクラスター化して視覚化します。
結果を解釈するときは、データQCに注目することが重要です。 Basepair のシングルセルRNA-seq レポートには、検出された遺伝子/特徴、UMI 数、ミトコンドリア比率などの細胞ごとのQC指標が含まれています。 クラスタリングおよび視覚化ツールを使用すると、クラスター全体の遺伝子の発現を視覚化できます。
BasepairのシングルセルRNA-seq パイプラインは、バーコード抽出に UMI ツール、アライメントに STAR ツール、QC に fastp、クラスタリングと視覚化に Seurat を使用します。 すべてのパイプラインは、引用度の高い、査読済みのツールを使用して構築されています。 さらに、公開されているデータセットを使用してすべてのパイプラインを検証しています。
Basepair には、例えば、治療グループと対照グループの両方に複数の複製をアップロードできる「Single cell RNA-seqintegrate」と呼ばれるパイプラインがあります。 Basepairはすべてのサンプルを個別に分析し、これらのサンプルをカウントマトリックスに組み合わせて、2 つのグループ間で発現の異なるクラスターを表示します。
Basepair は、パイプラインで使用する公開ツールの推奨デフォルト値に基づいてパラメーターを設定します。 これらのパラメーターはほとんどのデータセットで機能し、ほとんどのユーザーはこれらのパラメーターを変更する必要はありません。
主要な結果を他のデータセットまたは分析で確認することを常にお勧めします。 理想的には、さまざまな種類の技術 (分子アッセイなど) を使用します。 すべてのバイオインフォマティクス ツールにはある程度の不正確性があります。 結論を裏付ける複数の並行した結果セットがあることは常に好ましいといえます。
nUMI と nGenes の関係を明示的にチェックすることはありませんが、nUMI または nGenes が高すぎる、または低すぎる遺伝子を削除するためのしきい値を提供します。 Basepairが受け取るさまざまなデータセットに対処するために、nUMI と nGenes の中央値から 標準偏差で+/- 3離れたセルを個別に削除するという単純なアプローチを使用します。 これらのデフォルトのしきい値は独自のしきい値でオーバーライドできます。
遺伝子発現はまず各細胞内の総発現を考慮して正規化され、次に対数変換が適用されます。 次に、下流の教師なし解析 (PCA、t-SNE など) 用の遺伝子を選択するために、分散安定化変換 (VST) を使用して発現値の不均一分散性を補正します。 また、総 UMI 数、検出された総遺伝子、ミトコンドリアにマッピングされている読み取りの割合の細胞ごとの測定値を回帰する方法も適用します。
はい、遺伝子を検索するか、表の右側にある虫眼鏡をクリックすることができます。 これにより、遺伝子の発現レベルで各セルを色付けすることにより、t-SNE プロットが更新されます。 バイオリン プロットは、t-SNE プロットに示されているクラスターによって分割された遺伝子の発現もプロットします。
します(良い意味で)。 リードフィルタリングは、品質の低い細胞(死滅または瀕死の細胞など)や周囲の RNA に由来するリードを除去するのに役立ちます。これらはいずれも発現差に悪影響を与える可能性があります。 フィルタリングは UMI ツールを使用して実行され、最初に同じバーコードを持つ読み取りをグループ化します (各グループは理想的には 1 つのセルを表します)。
はい。「Copy data to GEO」という自動パイプラインがあり、提出のために必要なファイルを NCBI に自動的に送信します。
最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。