
BasepairのRNA-Seq
fastqがあれば、Basepairを使うことで、RNA-Seqの解析を迅速に行うことができます。一気にデータQCやアライメントからDEGや視覚化まで完了します。
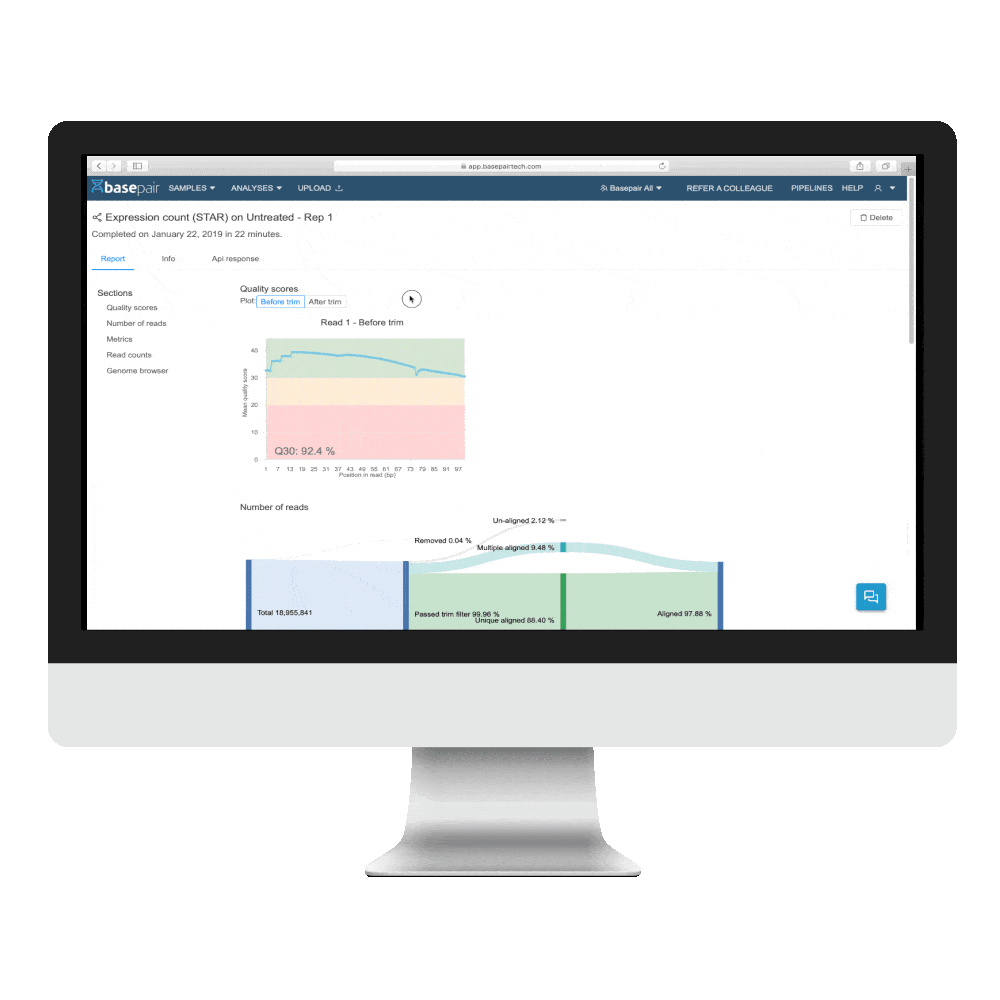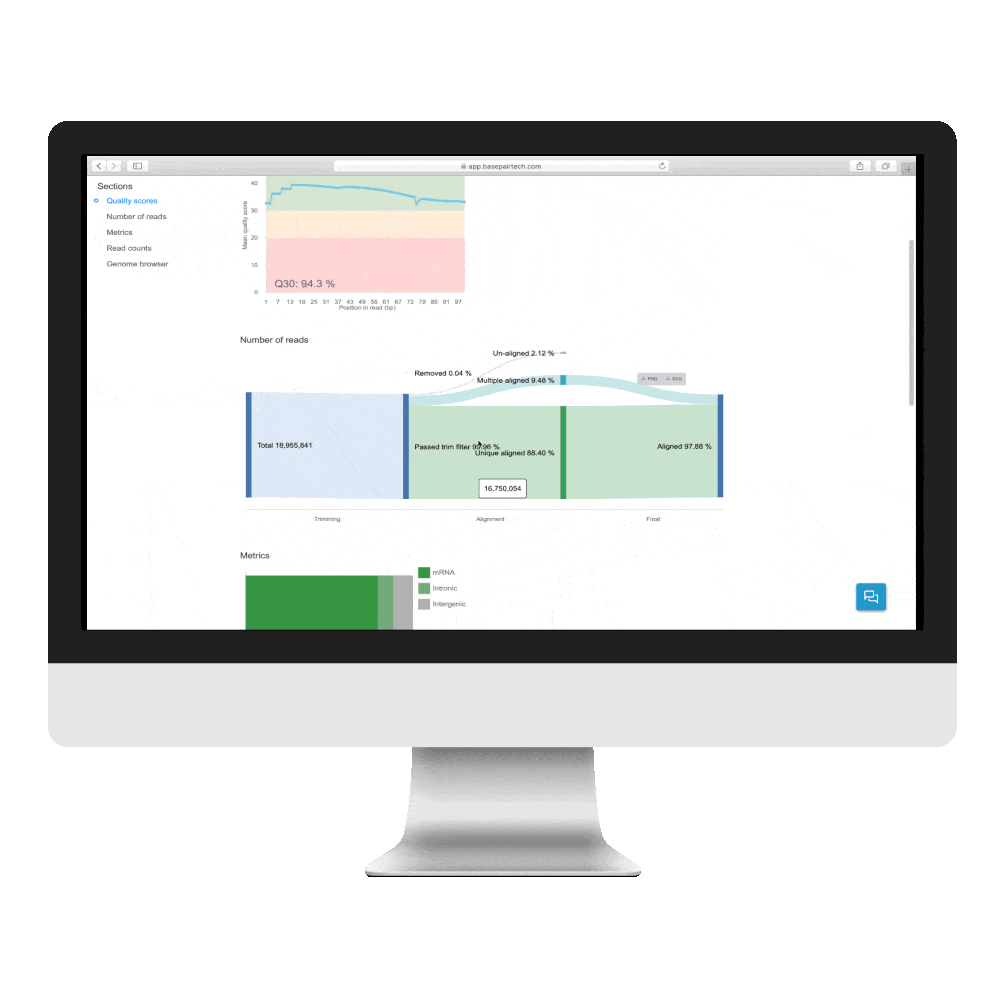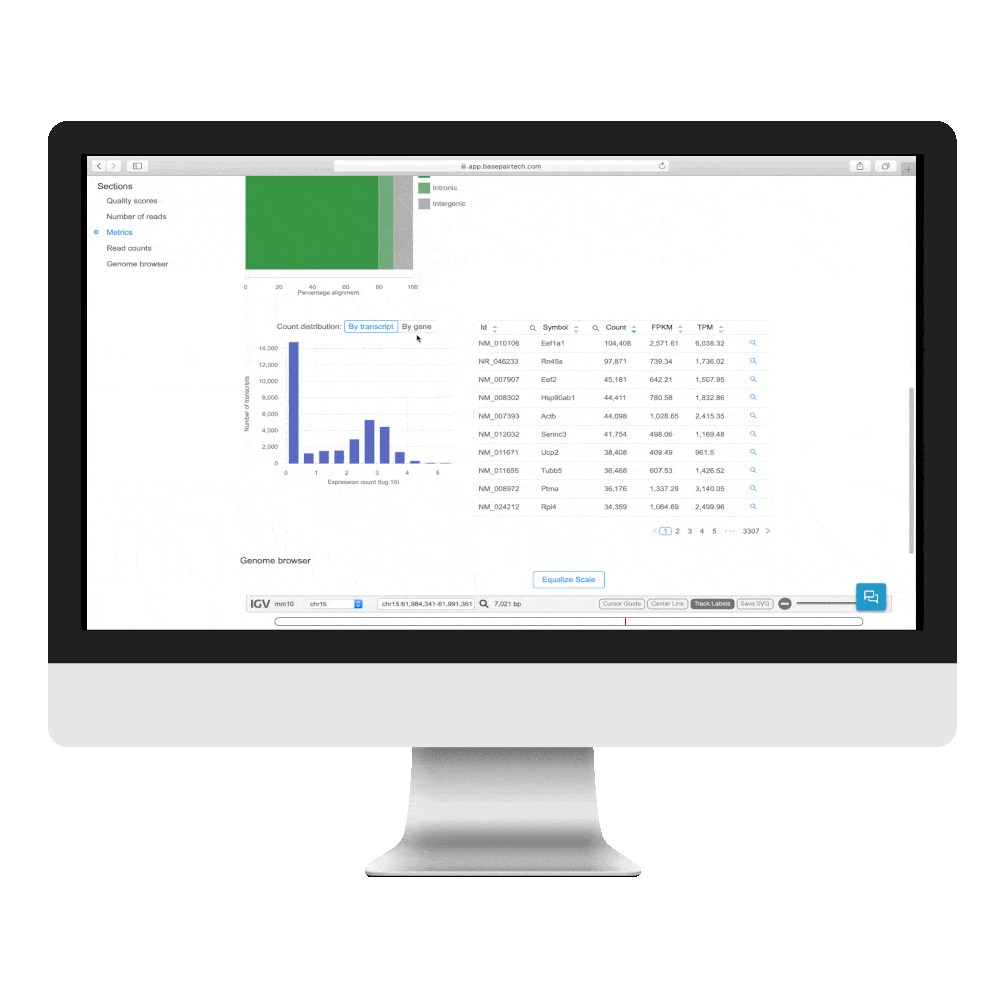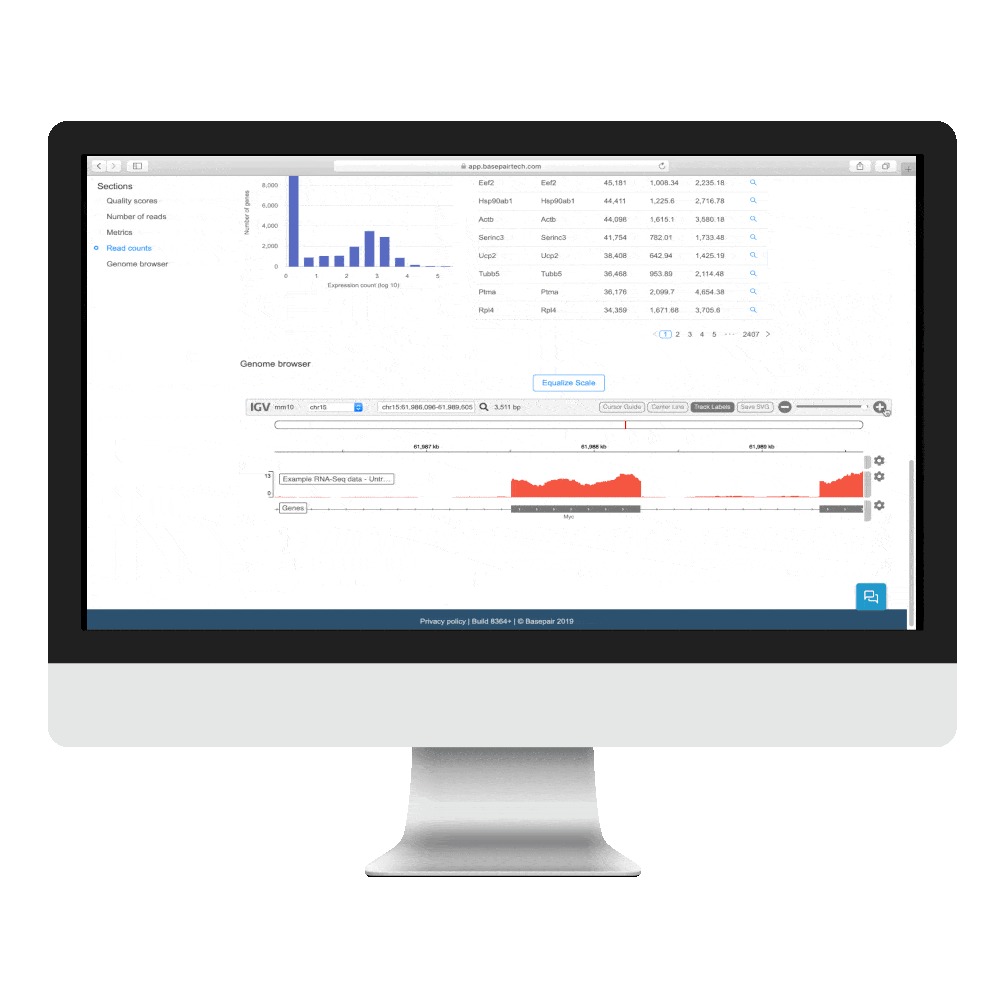
fastqをアップロードするだけでDEGまで一気に完了
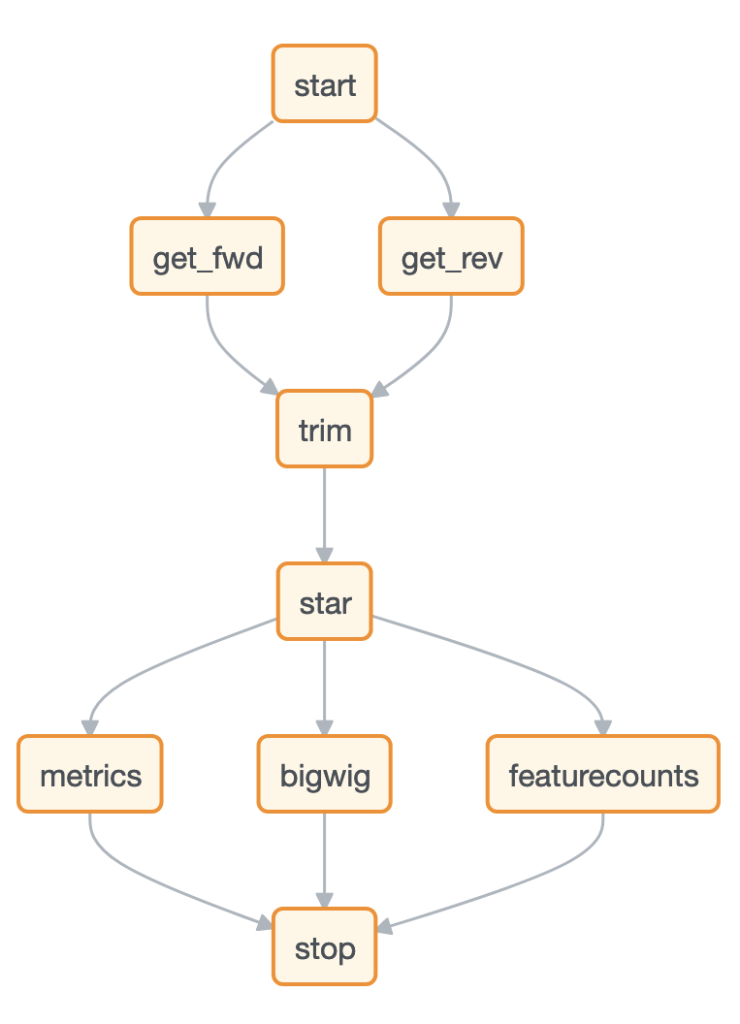
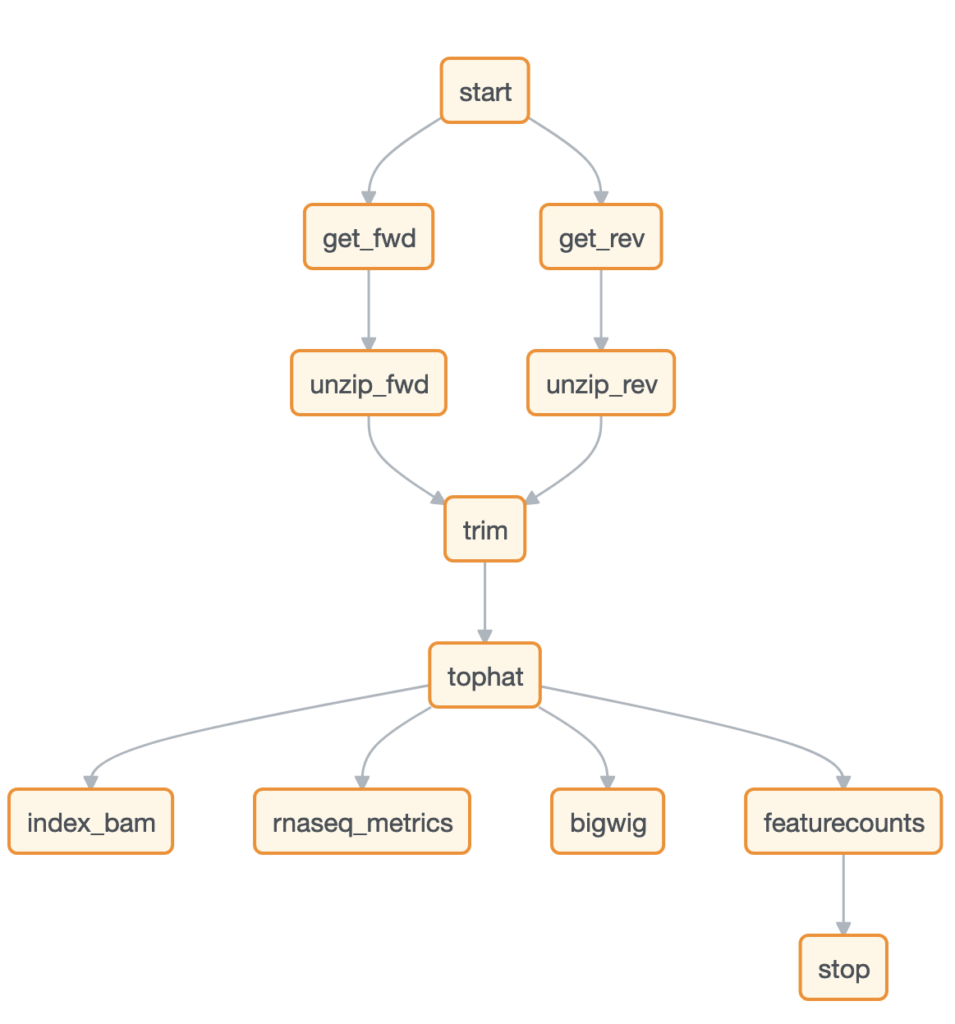
BasepairのRNA-Seqでは、fastq(.gz)をアップロードするだけです。解析を開始すると、必要なワークフローを実行し、データQC、トリミングからDEG(遺伝子発現差解析)まで自動的に一気に完了します。
ファイルフォーマットを気にしたり、パイプライン同士を繋げたりする必要はありません。
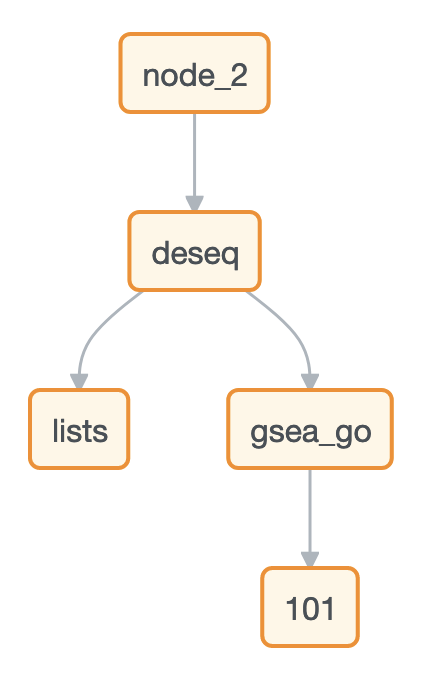
発現量カウントにはSTARとTophatを用意しており、選ぶことができます。また、DEG(遺伝子発現差解析)には、DESeq2を使用しており、2グループ間、あるいは3グループ間以上の比較も可能です。DESeq2を起動した際に、発現カウントがない場合は、自動的に発現カウントパイプラインが実行されます。
GSEA(エンリッチメント解析)も含まれており、グラフが生成されます。
パイプライン
すべてのパイプラインは、引用度の高い、査読済みのツールで構築されています。
- DESeq2:2グループ間、3グループ間以上での遺伝子発現量の比較
- STAR:QC、アライメント、発現量カウント
- Tophat:QC、アライメント、発現量カウント
- deFuse:融合遺伝子イベント
- Trinity:de novoアセンブリー
- Cufflinks:アセンブルと遺伝子発現量の比較
- Cuffdiff:アイソフォームレベルでの発現量解析
すべてのパイプラインは、プラットフォーム内で確認できます。(ログインが必要です)
遺伝子発現量の定量化
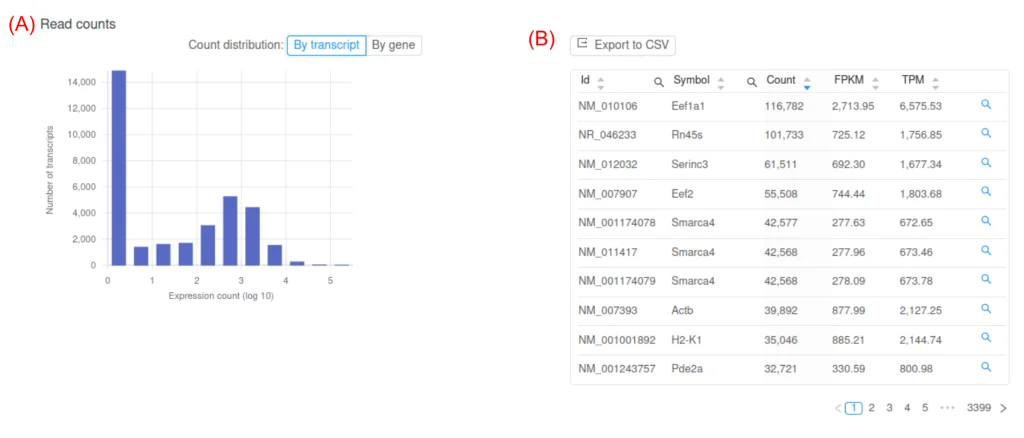
RNA-Seqで得られた発現量カウントはグラフと表で表示されます。発現量は、rawカウント、FPMK、TPMで表示され、CSVでのダウンロードが可能です。
遺伝子発現量の比較
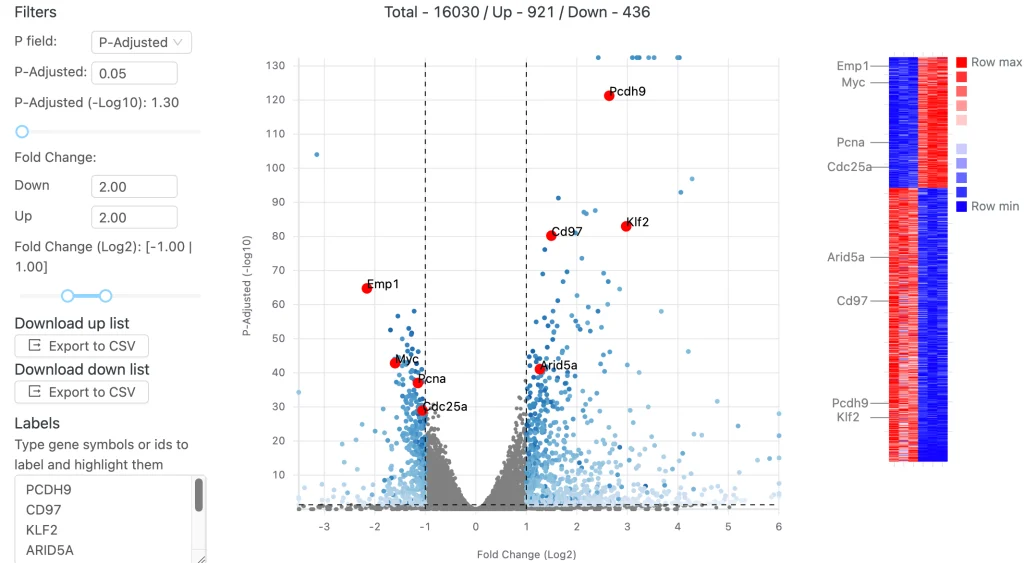
RNA-Seqで得られた発現量の比較をする場合はDESeq2を用いて、発現量のlog2とp値で表現されたボルケーノプロットを作成することができます。発現量とp値はそれぞれゲージを動かすことで、絞り込みを行うことができます。発現量の表とも連動しており、目的の遺伝子のみを表示したり、ラベルを付与することができます。
ヒートマップが練度しており、絞り込みを行うと自動的にヒートマップも更新されます。
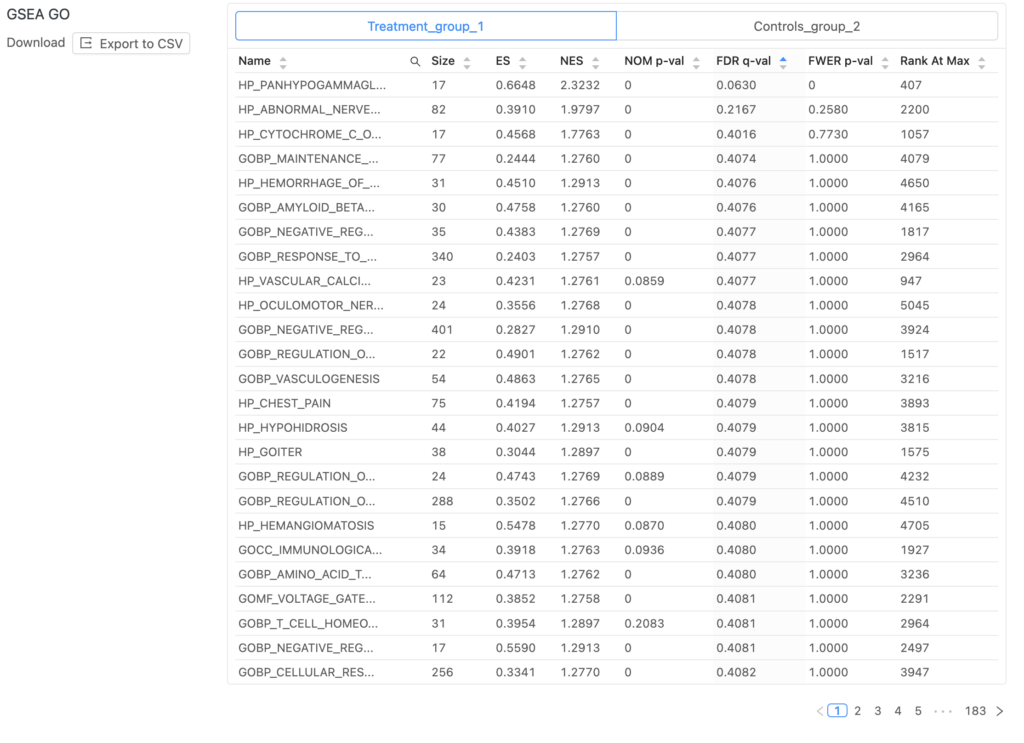
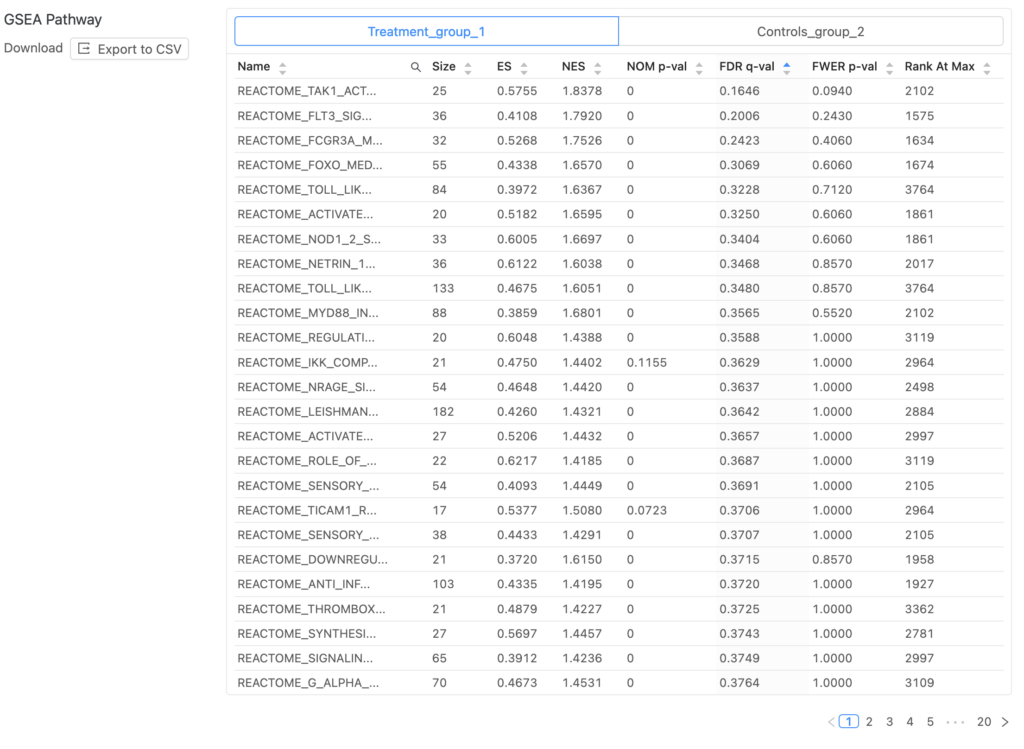
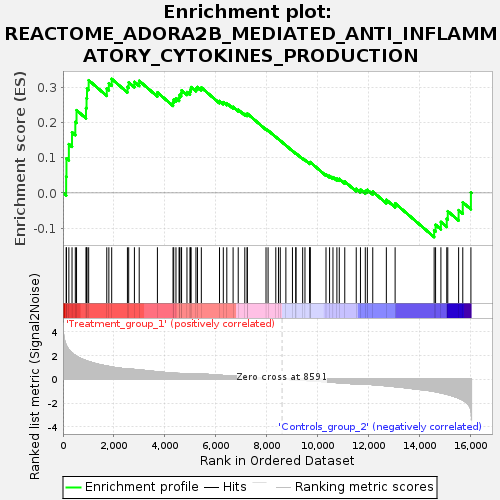
GSEA(エンリッチメント解析)
RNA-Seqで得られたデータを解釈する際によく行われるGSEA(エンリッチメント解析)も用意しています。DESeq2は実行れるとあわせて自動的に実行されます。GO(Gene Ontology)とPathwayの遺伝子セットを用いたGSEA(Gene Set Enrichment Analysis)も実装されています。それぞれにエンリッチメントプロット(Enrichment plot)が生成されます※。
※ 多量のファイルが生成されるため、zipでのダウンロードになります。
関連記事
-
 TechBlog
TechBlog
BasepairのGene fusion finding (deFuse) パイプライン
deFuseパイプラインは遺伝子融合を検出するために使用されます。このパイプラインはまずリードをリファレンスゲノムにアライメントします。次に、融合している可能性のある遺伝子を検索します。最後にフィルタリングを行い、偽陽性 […]-
iwai-chem
-
-
TechBlog
BasepairでのCuffdiffパイプライン
Cuffdiffパイプラインは、サンプルグループ間のアイソフォームレベルの発現変動解析を実行するために使用されます。Cuffdiff パイプラインが実行されると、最初に Expression coun […]-
-
-
TechBlog
BasepairでのCufflinksパイプライン
Cufflinksパイプラインを実行すると、最初にExpression count (STAR)パイプラインが自動的に実行されます。このCufflinksパイプラインは、STARパイプラインでアラインメントされたリードを […]-
-
-
 Bioinformatics
Bioinformatics
なぜインタラクティブレポートなのか?
RNA-Seqのデータ解析は、エクセルやRと格闘したり、受注会社に依頼したりすることが多いと思います。これには、時間とコストがかかります。インタラクティブレポートを使うと、大きく時間をコストを圧縮することができます。 R […]-
hideki
-
-
TechBlog
Basepairでパスウェイ解析(Reactome)
パスウェイ解析とは Basepairではパスウェイ解析も行うことができます。あらかじめ定義された遺伝子や遺伝子産物の位置、遺伝子同士の相互作用などの情報を含んだモデルをもとに行うため、より生物学的な議論を行う […]-
elisa
-
-
 TechBlog
TechBlog
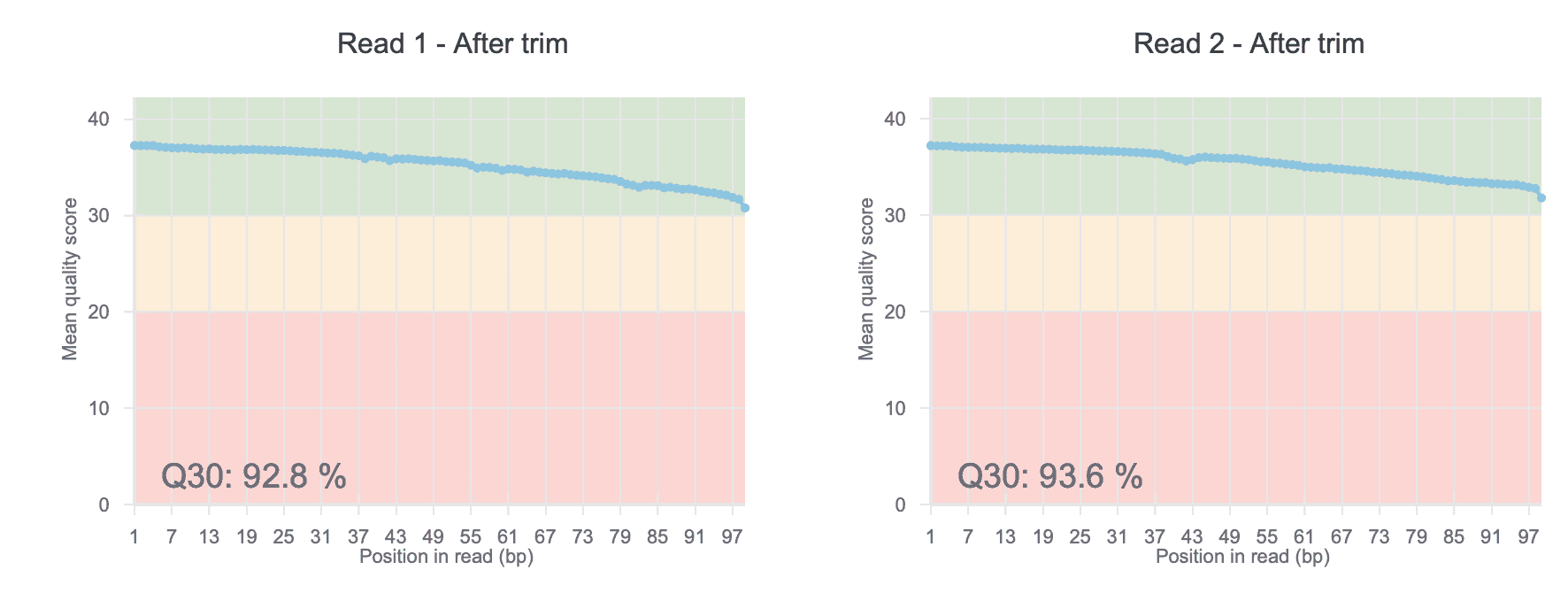
FASTQファイルの品質評価と前処理
なぜFASTQの品質が重要か FASTQの品質は、下流の解析の品質に影響します。 FASTQデータには、低品質リードが含まれることがあります。多くの場合、これはシーケンシングエラーやPCRバイアスなどの要因によって生じま […]-
-
-
Bioinformatics
KEGGパスウェイ解析
BasepairのRNA-SeqのDESeq2パイプラインは、発現変動解析、Gene Ontology(GO)エンリッチメント解析、Reactomeパスウェイエンリッチメント解析と統合されています(図1)。また、必要に応 […]-
-
-
Bioinformatics
Basepair上でエンリッチメント解析 (GSEA/Gene set enrichment analysis)
エンリッチメント解析(GSEA)はRNA-Seqの際によく行われます。とはいえ、Rなどプログラミングを使用して実行するのは高いハードルがあります。また実行時間を要します。Basepairでは数回クリックするだけでGSEA […]-
-
-
 TechBlog
TechBlog
バイオインフォマティクスのコスパとタイパ
Basepairは、fastqなど生データがあれば、NGSデータの解析ができます。一方で、自分で解析することが重要・必要なケースも少なからずあると思います。とはいえ、自分でやるべきか、判断に迷うこともあると思います。そこ […]-
-
Basepairは、査読ジャーナルにも使われています。





最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。