
A Gentle Introduction to RNA-Seq Analysis
RNA-seq は、前例のないレベルの感度でトランスクリプトーム全体を検査するテクノロジーです。 RNA-seq には、発現の定量化、新規遺伝子や遺伝子アイソフォームの発見、発現差、その他多くの種類の機能解析に至るまで、幅広い用途があります [1]。 したがって、研究者は RNA-seq を使用して、最も切実な質問の多くに答えることができます。 新しいがんのサブタイプの発見、これまで配列決定されたことのない種のトランスクリプトームの発見、組織特異的な遺伝子発現の解明、その他多くの応用が可能です。 この目まぐるしい RNA-seq アプリケーションの配列のため、RNA-seq データを分析する際の主な手順を優しく紹介したいと思います。
- 品質管理とトリミング
- 読み取りアライメント
- 式を定量化する
- 差次的発現
品質管理とトリミング
最初のステップとして、低品質のリードとアダプター配列をデータから削除する必要があります ( トリミング と呼ばれるステップ)。 アダプターはシーケンス用にサンプルを準備するために使用される短い配列であり、分析前に削除する必要があります。 Basepair では、トリミングが読み取り品質に及ぼす影響は、通常、図 1 のような図で示されます。しかし、品質はどのように測定されるのでしょうか? シーケンスマシンは、シーケンスされた各塩基対の品質スコア (Q 値と呼ばれます) を出力します。 Q 値が高いほど、データが良好であることを意味します。 トリミングは、読み取りの低品質部分、または必要に応じて読み取り全体を削除するため、重要です。 これにより、下流の分析に役立つデータの量が増加します。 トリミングがデータ品質にどのような影響を与えるかについての詳細な概要については、 ブログ投稿を参照してください。 このテーマに関する

読み取りアライメント
トリミング後、リードは通常、参照ゲノムまたはトランスクリプトームのいずれかにアライメントされます。 アライメントは、リードがどの遺伝子に由来するかを判断するアルゴリズムです。 Basepair は、人気のある STAR ツールと TopHat ツールを提供します [2、3]。 あなたの種に参照がない場合のもう 1 つのオプションは、同じく Basepair [4] で提供されている Trinity を使用して独自のトランスクリプトームを組み立てることです。
調整が完了した後に注意すべき指標がいくつかあります。 理想的には、リードはゲノム内の 1 つの場所に一意にアラインメントされており、通常は 70% を超える一意にアラインメントされたリードを確認する必要があります。 ただし、一部のリードは複数の位置にアライメントされる場合があるため、リードがどの遺伝子に由来し、削除されるかは不明です。 アライメントは、SAM 形式 (シーケンス アライメント マップ) でデータを出力します。その後、圧縮形式 (BAM) に変換され、さらなるダウンストリーム分析に使用されます。 Basepair は、合計開始リード数、トリミングされたリード数、アライメントされたリード数までのリード処理ステップを視覚化します (図 2)。
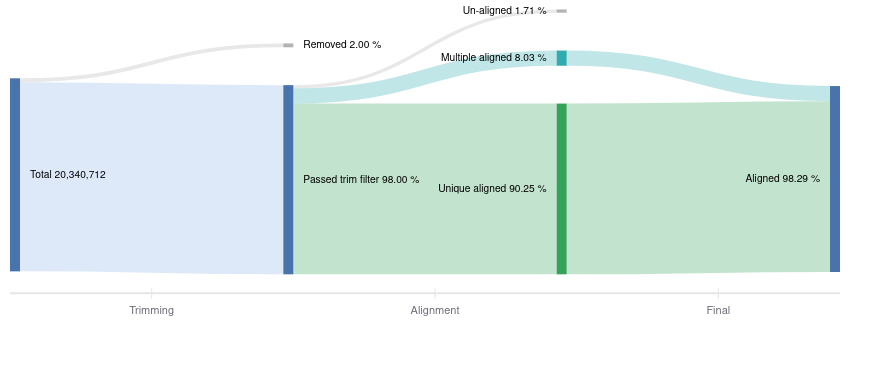
図 2: Basepair の代表的な Sankey プロット。生データからトリミングとアライメントまでの読み取り量を示します。
発現の定量化
次のステップは、遺伝子発現を定量することです。 各遺伝子に一致するリードの数は、HTSeq-count や featureCounts などのプログラムを使用して定量化されます。 ただし、生のリード数は、サンプルと遺伝子間の発現を比較するのには適切ではありません。 生のリード数には、遺伝子の長さや各サンプルのリードの合計数などのバイアスは考慮されていません。 FPKM (マップされたリード数 100 万件あたりのエクソン モデルのキロベースあたりの断片数) という測定値は、一般的なサンプル内正規化方法です [2]。 Basepair RNA-seq 解析パイプラインは、raw カウント、FPKM、および別の一般的な正規化手法である TPM での発現を提供します (図 3)。
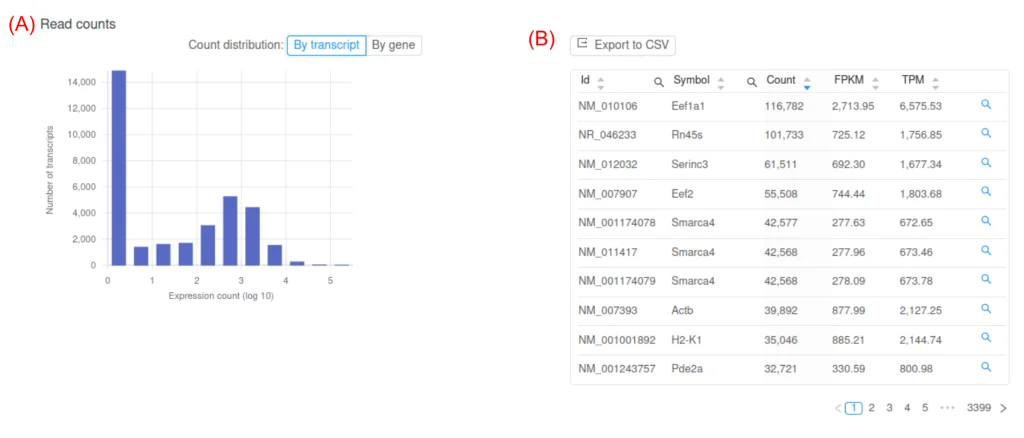
図 3: Basepair が提供する図と表の代表的なスクリーンショット。 (A) 対数変換された FPKM 値のヒストグラム。 上部には、遺伝子または転写産物の発現を切り替えるボタンがあります。 (B) 遺伝子ごとの発現値の対話型テーブル。並べ替えおよび検索できます。
遺伝子発現量比較
RNA-seq データのもう 1 つの一般的な分析は、2 つのサンプル グループ間の遺伝子発現の変化を統計的にテストする差次的発現解析(DEG、Differential Expression Gene)です。 たとえば、これは、治療条件と対照条件の間で変化する遺伝子を見つけるのに役立ちます。 Basepair は、差次的発現を実行するために非常に引用されている DESeq2 ツールを使用します。このツールは、遺伝子ごとの生の読み取り数を入力として受け取り、log2 倍数変化、p 値、および調整された p 値 (他の統計情報の中でも特に) を出力します [5]。 log2 倍率変化は遺伝子発現の変化の大きさを示し、p 値と調整された p 値は統計的有意性を評価します。 p 値と比較すると、調整された p 値は偽陽性が発生する可能性が低いため、検討するのに適した指標です。
Basepair を使用すると、差次的発現結果を対話的に調べることができます (図 4)。 さらに、より複雑な分析設定が必要な場合でも、Basepair が役立つ可能性があります。 Basepair は、2 つのグループの比較に加えて、3 つ以上のグループ、時間経過、入れ子条件の比較など、より複雑な実験セットアップに対応できます。
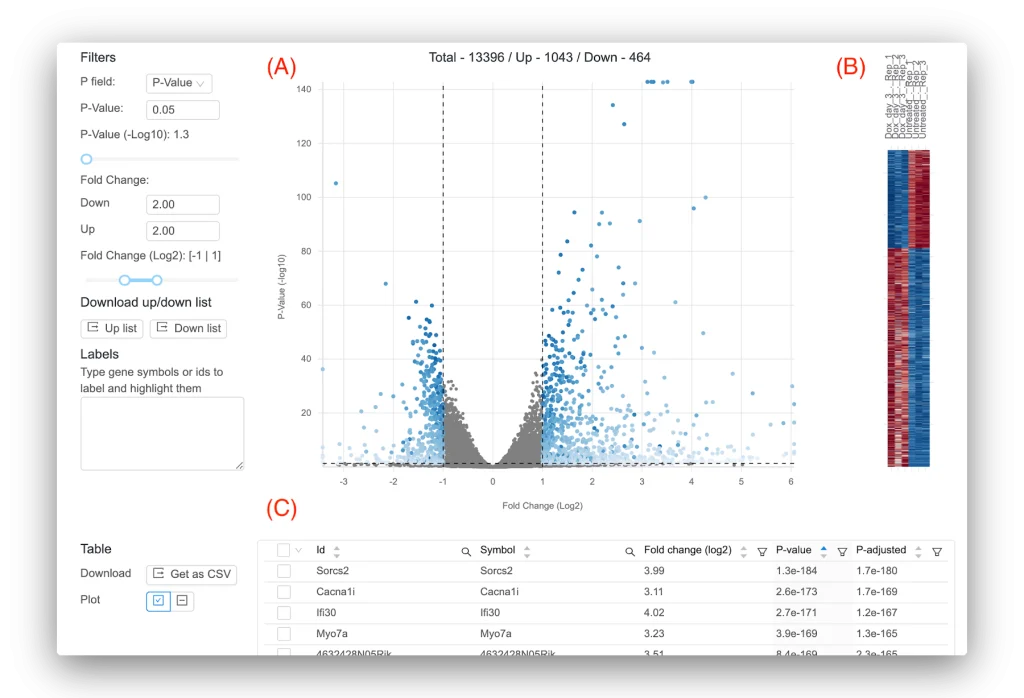
図 4: Basepair によって提供される代表的な結果。 (A) 各遺伝子の -log10(p 値) を log2 倍率変化に対してプロットするボルケーノ プロット。 重要な遺伝子は青色で表示されます。 左側のコントロールを使用して、重要性を判断するしきい値を設定できます。 (B) サンプル全体での重要な遺伝子の発現を示すヒートマップ。 (C) log2 倍率変化、p 値、および調整された p 値の遺伝子ごとの値を示す対話型の表。 列を並べ替えたりフィルターしたり、お気に入りの遺伝子を検索したりできます。
微分表現について詳しくは「 A Deep Dive Into Differential Expression」もご覧ください。
References
1. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Publishing Group. 2009 Jan;10(1):57–63.
2. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 Jan 1;29(1):15–21.
3. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. Oxford University Press; 2009 May 1;25(9):1105–11.
4. Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the 5. Trinity platform for reference generation and analysis. Nat Protoc. Nature Publishing Group; 2013 Aug;8(8):1494–512.
5. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. BioMed Central Ltd; 2014;15(12):550.