
Integrateパイプラインは、異なるシングルセルRNA-seqサンプルを比較するために使用します。ここでは、シングルセルRNA-seqパイプラインで生成された各サンプルのSeurat Objectをインプットとして使用します。まず、サンプル間のSeurat Objectを統合します。次に、統合されたデータにフィルターをかけ、正規化します。その後、クラスタリングと可視化が行われます。最後に、サンプル間の各クラスタのマーカー遺伝子が同定されます。
Integrateパイプラインは、シングルセルRNA-seqパイプラインからアウトプットされたSeurat Objectを必要とします。このため、Integrateパイプラインの実行を選択すると、選択されたすべてのサンプルにおいて、最初にsingle cell RNA-seqパイプラインが自動的に実行されます。ただし、すでにシングルセルRNA-seqパイプラインが実行されているサンプルについては、Integrateパイプラインの実行が選択されると、すぐにIntegrateパイプラインが開始されます。
パイプラインのワークフロー
図1にIntegrateパイプラインのワークフローを示します。
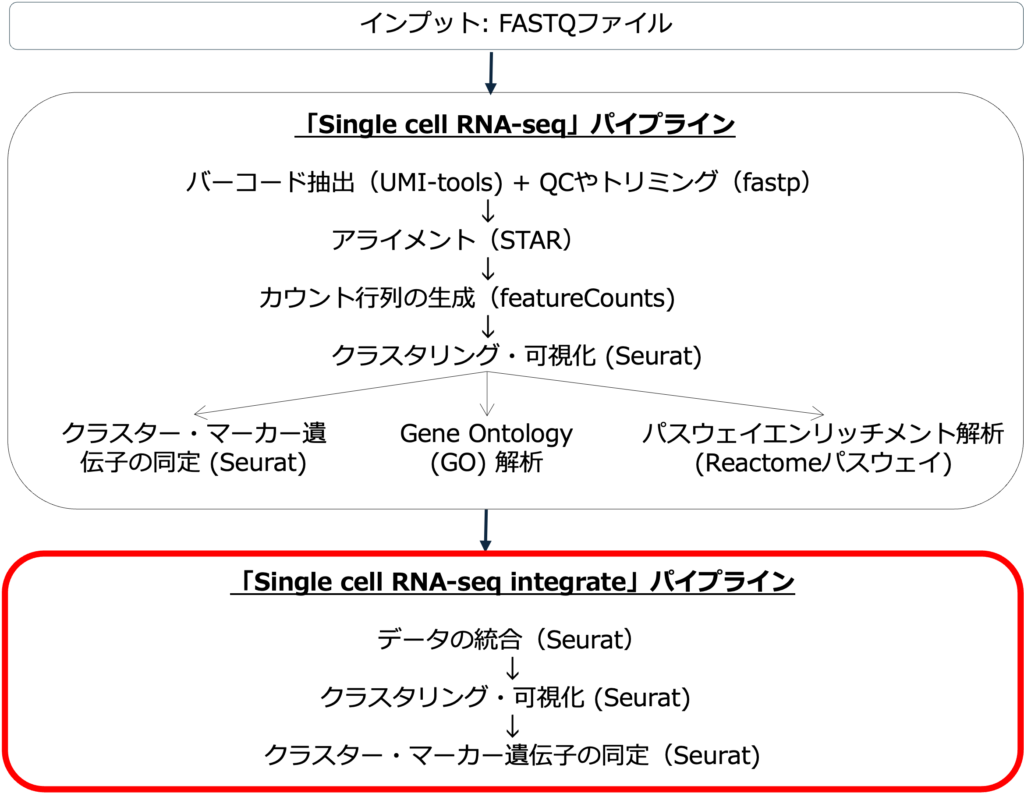
データの統合
Seurat ObjectsはSeuratによって統合されます。統合の目的は、生物学的変異を保持しつつ、サンプル間の技術的差異を整列させ補正することです。統合はデータセット間のアンカーを見つけることに基づいて行われます。アンカーとは、異なるデータセット間で類似した発現プロファイルを持つ細胞のことです。これらのアンカーは、異なるサンプル間の技術的なばらつき(例えば、ノイズ)を整列し、除去するために使用されます。ただし、技術的ばらつきの影響を受けている部分のみが調整されます。これにより、類似したプロファイルを持つ細胞がデータセット間で適切にアライメントされます。
サンプル間で類似した発現プロファイルを持つ細胞は統合時にアライメントされますが、サンプル間で差のある発現を持つ細胞はその差を保持します。従って、統合されたデータには、異なるサンプル間で共通の遺伝子とユニークな遺伝子の両方が含まれます。これにより、共通の細胞タイプと発現差のある遺伝子の両方を解析することができます(1)。
クラスタリングと可視化
統合後、Seuratを用いて統合データセットに対してクラスタリングを行います。これは、類似した発現プロファイルを持つ細胞をクラスターにグループ化するプロセスです。したがって、クラスターは異なるサンプル間で類似した遺伝子発現プロファイルを持つ細胞のグループを表します。
クラスタリングが完了すると、クラスターを低次元空間で可視化することができます。このプロセスをヴィジュアライゼーションと呼びます。これは細胞のタイプやクラスター間の関係を理解するのに役立ちます。また、サンプル間の生物学的差異を理解するのにも役立ちます(1)。
クラスタリングと可視化は両方、次元削減によって実行されます。
クラスタリング
Principal Component Analysis (PCA)によって行われます。PCAは次元削減の最初のステップです。
PCAは高次元の遺伝子発現データを、データ中の最大の分散を表せるより低い次元(principal components、PC)に還元します。言い換えると、PCAはデータの最大分散を表すPCを特定します。PCAで縮小されたデータは、可視化の入力として使用されます(1)。
可視化
データを可視化するために以下の手法を用います:
- t-SNE (t-distributed stochastic neighbor embedding)
- UMAP (Uniform Manifold Approximation and Projection)
どちらの手法も異なるサンプルの細胞間の関係を可視化するために使用されます。まず、PCAで縮小されたデータの細胞間の類似度が計算されます。次に、その類似性をより低い2次元または3次元空間に投影して可視化します。その結果、類似した発現プロファイルを持つ細胞は、プロットの中でより近くに配置され、緊密なクラスターを形成します。一方、類似していない細胞は離れて配置されます。
マーカー遺伝子の同定
同定されたクラスターはSeuratを用いてアノテーションされます。これはクラスター内のマーカー遺伝子を同定することによって行われます。マーカー遺伝子とは、特定のクラスターにおいて、他のクラスターと比較して有意な発現差を示す遺伝子のことです。マーカー遺伝子を同定することは、各クラスタの生物学的同一性を理解するのに有用です。
このパイプラインでは3種類のマーカー遺伝子の同定が行われます:
- クラスターマーカー遺伝子の同定
- 保存マーカー遺伝子の同定
- 条件間で差のあるマーカーの同定
クラスターマーカー遺伝子の同定
クラスター遺伝子マーカーは、サンプルグループ内の他のクラスターと比較して、特定の細胞クラスターで差次的に発現する遺伝子です。これらのマーカーを同定することで、サンプルグループ内の各クラスターの識別が容易になります。図2にこの解析のワークフローを示します。
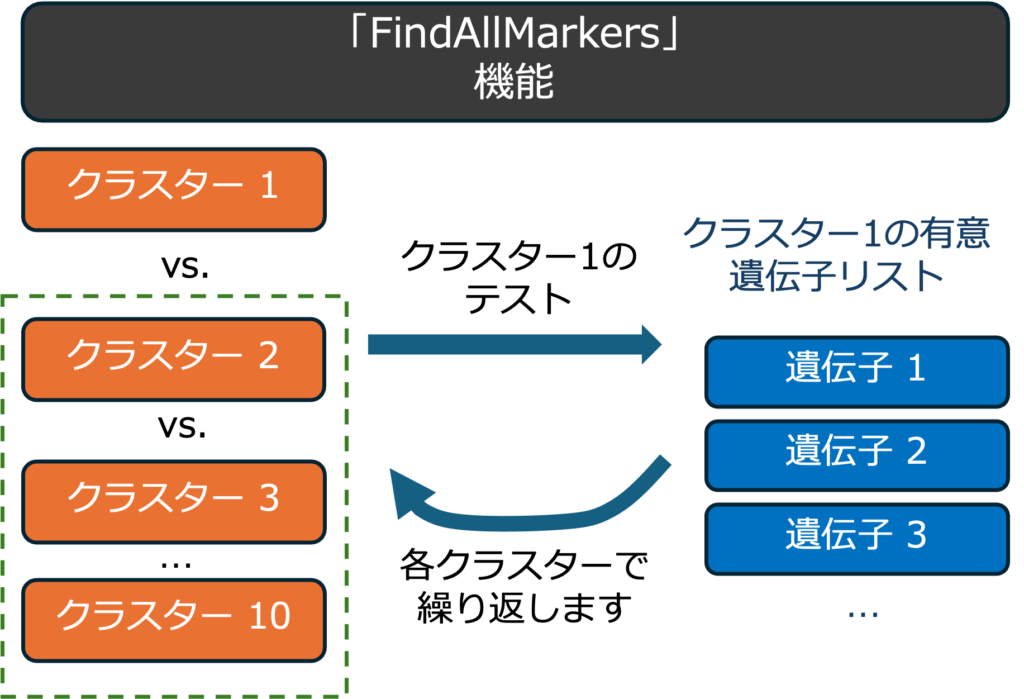
まず、あるクラスター(例えば、クラスター1)の遺伝子の発現を、他のすべてのクラスター(例えばクラスター2、3など)と比較するために、差次的発現解析を行います(図2a)。次に、Seuratの「FindAllMarkers」機能を使って、その特定のクラスター(例えば、クラスター1)のマーカー遺伝子を同定します(図2b)。このプロセスを他のすべてのクラスターにおいても繰り返します(図2c)。
保存マーカー遺伝子の同定
保存遺伝子マーカーは、異なるサンプルグループ間で特定のクラスターで発現している差次的発現遺伝子です。したがって、保存遺伝子マーカーは、異なるサンプルグループ間で特定のクラスターに保存されている遺伝子です。この解析は、異なるサンプルグループ間で保存されている細胞タイプマーカーを見つけるのに有用です。図3にこの解析のワークフローを示します。
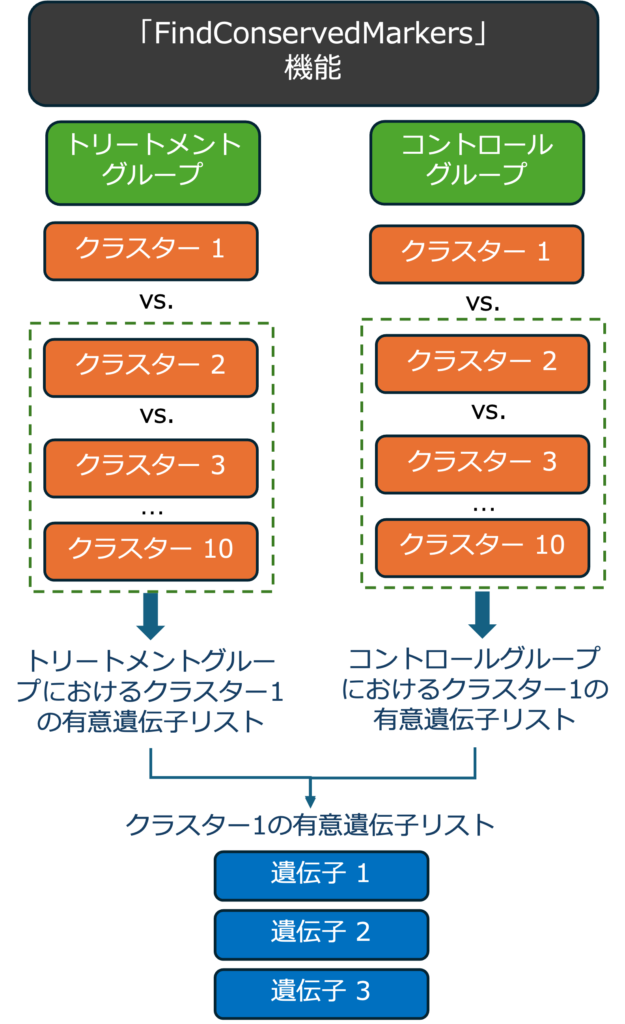
まず、あるクラスター(例えば、クラスター1)内の遺伝子の発現を、1つのサンプルグループ内の他のすべてのクラスター(クラスター2、3など)と比較するために、差次的発現解析を行います。同じステップをもう一方のサンプルグループに対しても行います(図3a)。次に、Seuratの「FindConservedMarkers」機能を用いて、各サンプルグループ内の特定のクラスター(例えば、クラスター1)に含まれるマーカー遺伝子を同定します。このステップを他のすべてのクラスターについて繰り返します(図3b)。
したがって、各サンプルグループの各クラスタについて、1つの有意な遺伝子リストが存在することになります。次に、同定されたマーカー遺伝子を異なるサンプルグループ間で結合します。異なるサンプルグループ間でクラスターに保存されている遺伝子マーカーがアウトプットされます(図3c)。
条件間で差のあるマーカーの同定
この解析の目的は、異なるサンプルグループ間で特定のクラスター内の遺伝子の発現を比較し、差次的な遺伝子マーカーを同定することです。この解析は、特定の遺伝子が様々な生物学的状態(例えば、治療対コントロール)にどのように反応するかを理解するために重要です。図4にこの解析のワークフローを示します。
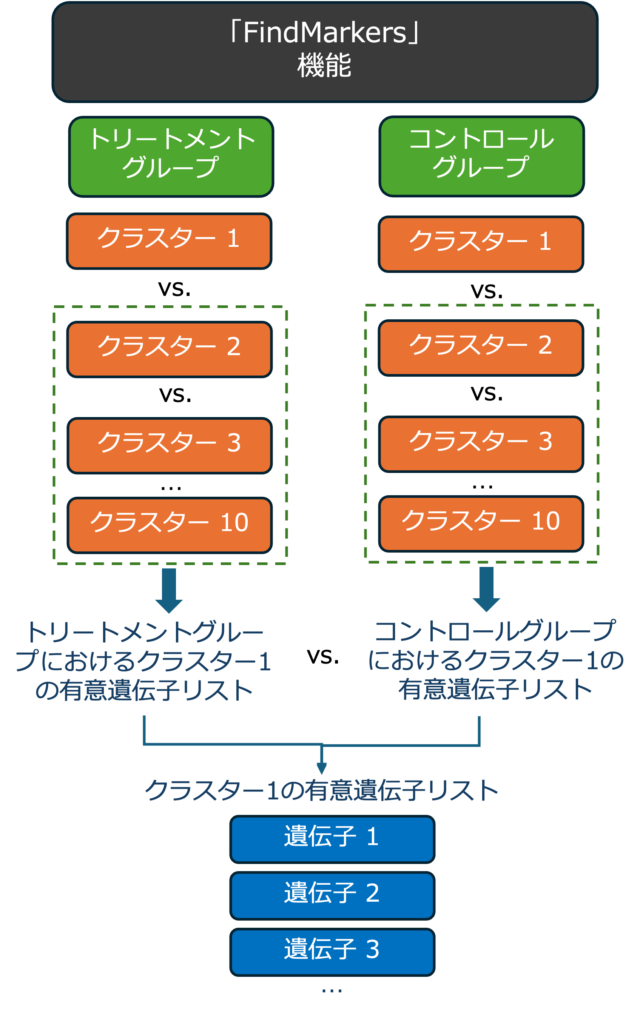
まず、1つのクラスター(例えば、クラスター1)の遺伝子の発現を、1つのサンプルグループ内の他のすべてのクラスター(クラスター2、3など)と比較するために、差次的発現解析を実行します。同じステップをもう一方のサンプルグループに対しても行います(図4a)。次に、Seuratの「FindMarkers」機能を用いて、各サンプルグループ内の特定のクラスター(例えば、クラスター1)内のマーカー遺伝子を同定します。このステップを他のすべてのクラスターについて繰り返します(図4b)。
したがって、すべてのサンプルグループのすべてのクラスターについて、1つの有意な遺伝子リストが存在することになります。次に、同定されたマーカー遺伝子を異なるサンプルグループ間で比較します。異なるサンプルグループ間のクラスターで差次的に発現している遺伝子マーカーがアウトプットされます(図4c)。
結果(「Report」タブ)
結果は「Report」タブ(図5の赤枠)にあります。
Summary
図6では、フィルタリング後の統合データにおける細胞と遺伝子・フィーチャーのサマリーを示します。このプロットは、クラスタリングとマーカー遺伝子の同定に進む前に、フィルタリングされた統合データの品質を評価するのに有用です。
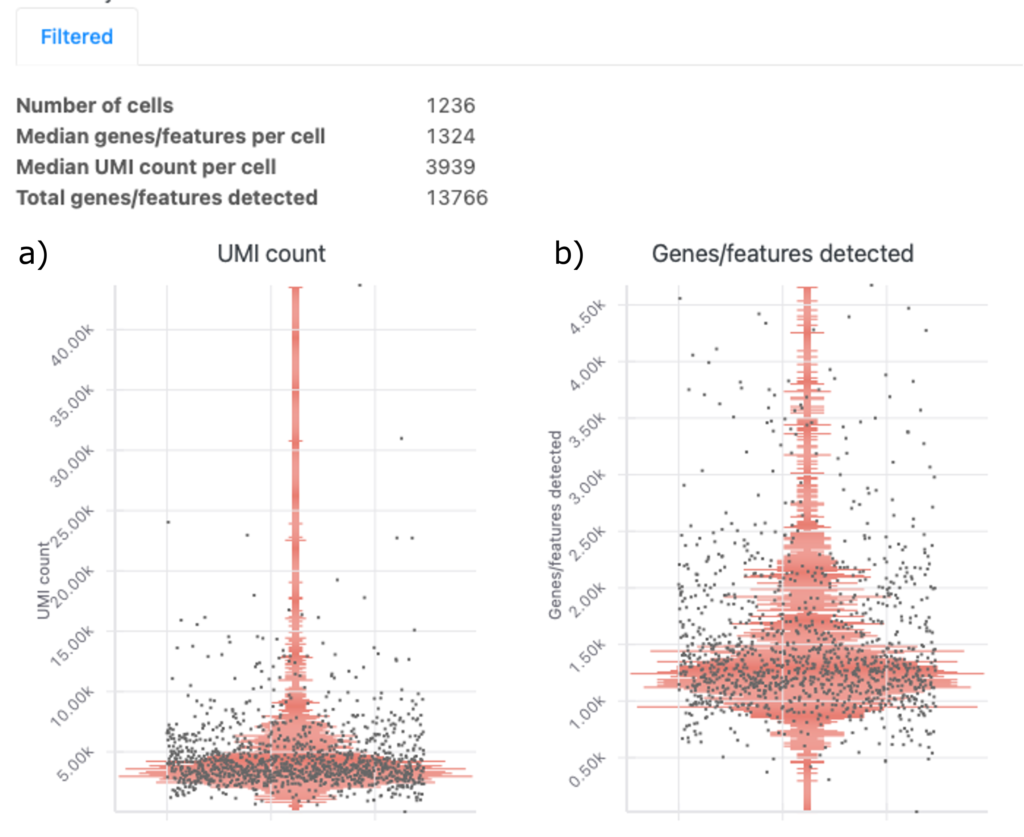
図3上段の用語
| 用語 | 定義 |
| Number of cells | 個々の細胞の総数 |
| Median genes/features per cell | 各細胞で転写産物が捕捉されたユニークな遺伝子/フィーチャー数の中央値 |
| Median UMI count per cell | 細胞あたり検出された転写産物の中央値 |
| Total genes/features detected | 全細胞で検出された遺伝子・フィーチャーの総数 |
図6aでは、UMIカウント分布プロットを示しています。この例では、X軸は統合データの細胞を表します。そして、Y軸は細胞あたりのUMIカウント(~40,000まで)を表します。それぞれの黒い点は個々の細胞を表します。赤い線はUMIカウントの一般的な密度を示します。大半の細胞のUMIカウントは、中央値(3939 UMI/細胞)付近でした。
遺伝子・フィーチャー分布プロットを図6bに示します。ここでは、X軸は細胞を表します。一方、Y軸は細胞あたり検出された遺伝子・フィーチャー数(最大~4500)を表します。図6aのように、黒い点は細胞です。また、赤い線は密度を表します。細胞あたり検出された遺伝子・フィーチャーの中央値は1,324でした。
Scatterplot
図7では、クラスタリング中に生成されたt-SNE、UMAP、PCAプロットを示しています。上部のタブ(図7青枠)をクリックすることで違うプロットを表示できます。
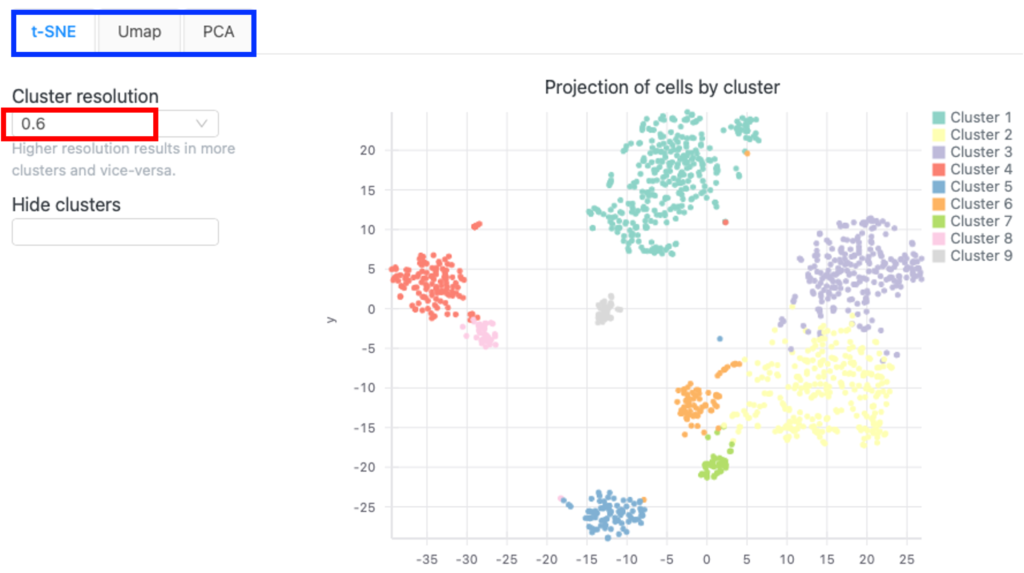
図7では、軸は高次元空間における局所的な類似性を保持する方法で、細胞の2次元投影を表しています。これらの軸の実際の単位には生物学的な解釈はありません。しかし、点間の相対的な距離には意味があります。
各ドットは1つの細胞を表します。互いに近い細胞は、似たような遺伝子発現プロファイルを持ちます。したがって、それらはクラスターにグループ化できます。一方、離れている細胞はより明確な発現パターンを持ちます。
全てのクラスターは異なる色で示されています。各クラスター内の細胞の決定はクラスタリングアルゴリズムに依存します。アルゴリズムはクラスタ分解能に影響されます(図7の赤枠)。クラスタ分解能は、アルゴリズムを適用する際に、細胞のクラスターをどの程度明瞭に、あるいは細かくするかを制御するパラメータです。
図7では、解像度を0.6として、細胞を9つのクラスターにグループ化しています。解像度の値が低いと、より少ない、より広いクラスターになります。逆に、解像度の値が高いほど、より多く、より細かいクラスターになります。
統合データにおけるクラスタの意味
クラスターは以下を表すことができます:
1) 異なる細胞タイプ
2) 異なる細胞の状態:
– 例えば、発生の異なる段階にある細胞は、別個のクラスターを形成します。
3) 生物学的特徴の共有:
– 統合されたデータでは、同じクラスターに入る異なるサンプルの細胞は、共通の生物学的特徴を共有している可能性が高いです。つまり、異なるサンプルグループに由来するにもかかわらず、類似した遺伝子発現プロファイルを持っています。
選択遺伝子のt-SNEプロットとバイオリンプロットの表示
図8は選択遺伝子のa) t-SNEプロットとb) violinプロットです。
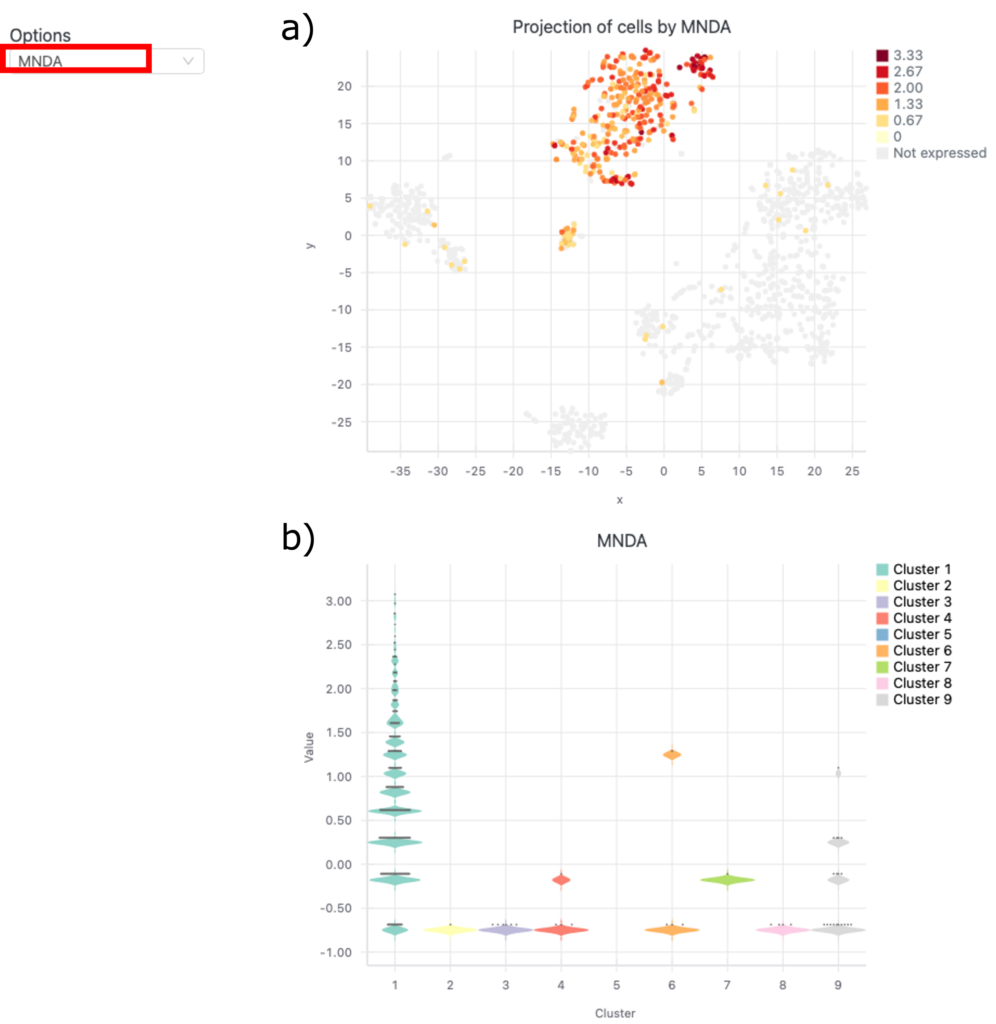
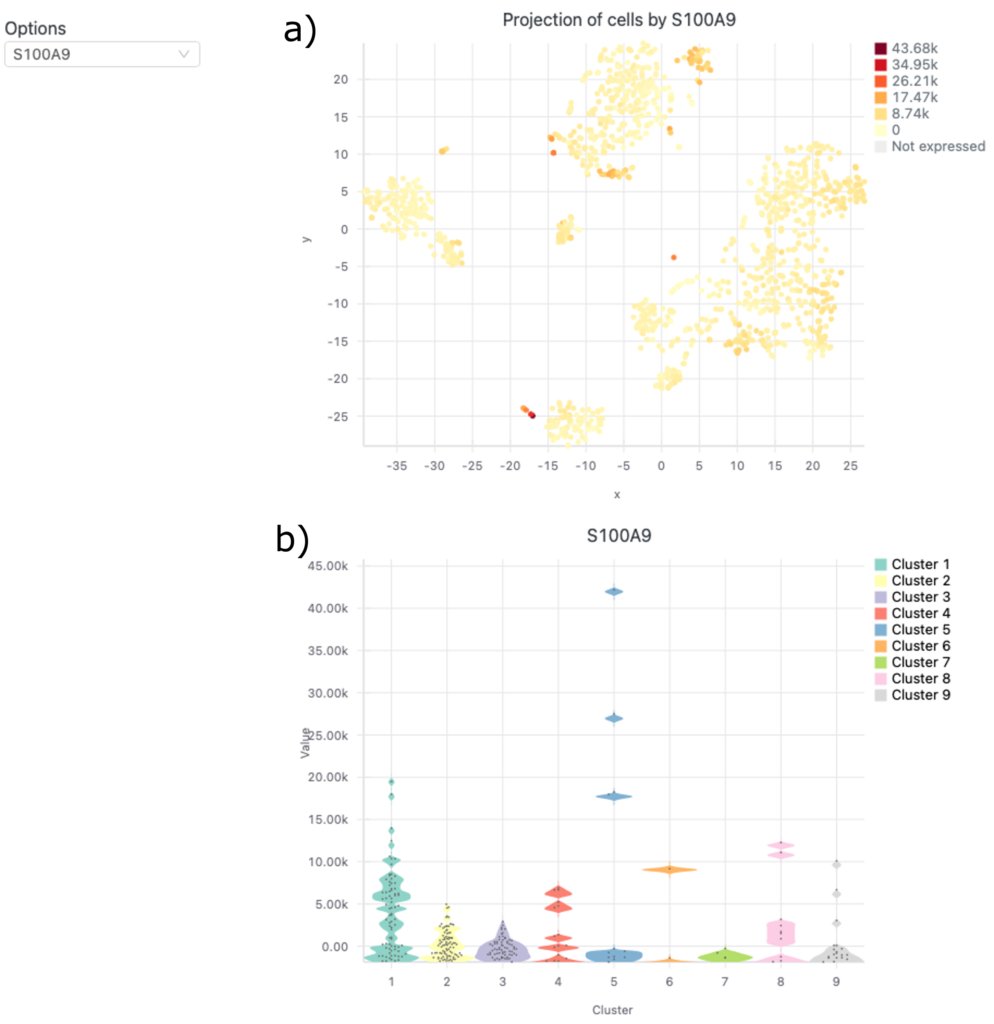
図7の赤枠で遺伝子を選択すると、a) t-SNEプロットとb) violinプロットを見ることができます(図8a)。これらは選択した遺伝子と分解能での発現分布を示しています。
図8aでは、x軸とy軸がt-SNE投影の2次元を表しています。各点は1つの細胞を表します。点の色は、選択されたMNDA遺伝子の発現レベルを示します。赤・高強度色はMNDA遺伝子の高発現を表します。一方、黄色・淡色はMNDA遺伝子の中程度の発現を表します。灰色は、MNDA遺伝子の無発現を表します。細胞の位置は、遺伝子発現パターンの類似性を反映しています。選択されたMNDA遺伝子の細胞の遺伝子発現は、クラスター1で最も高いです。逆に、他のクラスターはMNDA発現がほぼ見られませんでした。
図8bでは、異なる細胞クラスターにわたる選択されたMNDA遺伝子の発現が示されています。X軸は異なるクラスターを表します。Y軸はMNDA遺伝子の遺伝子発現レベルを示します。正の値は高い発現を表します。一方、負の値またはゼロの値は、発現が低い、または全くないことを示します。バイオリンプロットの結果は、t-SNEプロット(図8a)の結果と一致します。
Table
すべてのマーカー遺伝子を図9に示します。クラスターおよび保存マーカー遺伝子をそれぞれ図9aおよび9bに示します。異なるサンプルグループ間で発現が異なるマーカー遺伝子を図9cに示します。結果は、選択したクラスター(図9aの赤枠)と選択した分解能(図7の赤枠)に基づいています。
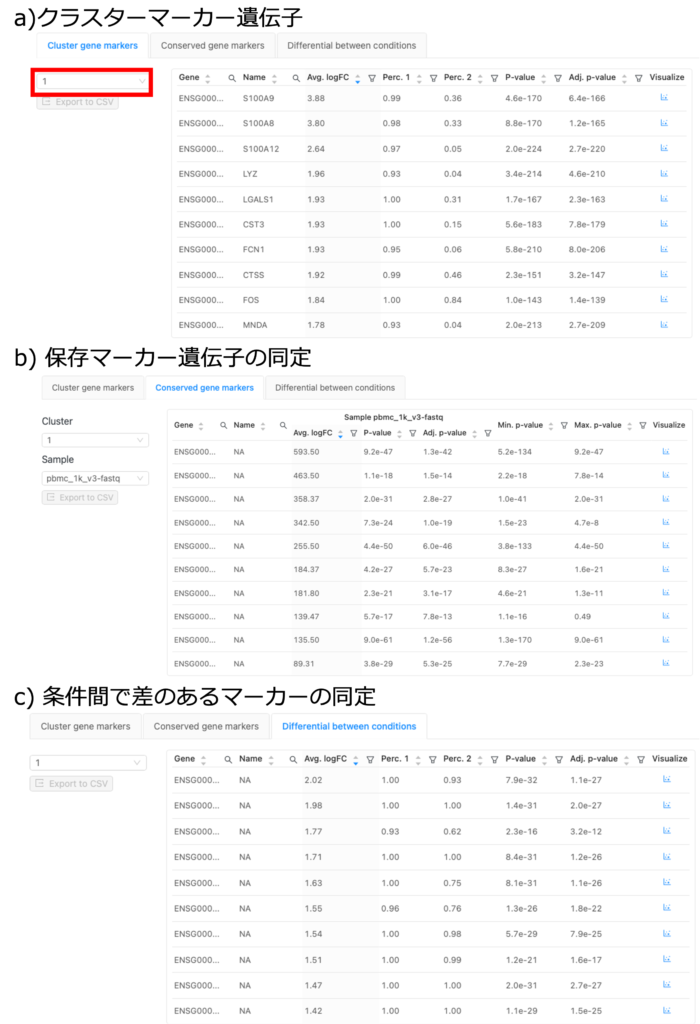
用語
| 用語 | 定義 |
| Gene | 同定されたマーカー遺伝子の記号 |
| Name | 同定されたマーカー遺伝子の名前 |
| Avg. logFC | Average log fold change。選択されたクラスターとそれ以外のクラスターにおけるこのマーカー遺伝子の遺伝子発現の差 FCが1:発現に変化なしFCが1以上:発現増加FC値<1:発現減少例えば、この場合選択されたクラスターはクラスター1です |
| Perc. 1 | 選択されたクラスターにおいて、マーカー遺伝子が発現している細胞の割合。 |
| Perc. 2 | マーカー遺伝子が発現している他の全てのクラスターにおける細胞の割合 |
| p-value | 選択されたクラスターと他のクラスターとの間の遺伝子発現の差の統計的有意性。p値が小さいほど有意差が大きいです |
| Adj. p-value | Adjusted p-value。Multiple hypothesis testingを考慮して補正されたp値。つまり、検出された偽陽性結果を補正する尺度 |
| Min. p-value | 異なるサンプル群間でマーカー遺伝子について観察された最小p値 |
| Max. p-value | 異なるサンプル群間でマーカー遺伝子に観察された最大p値 |
| Visualize | 選択した遺伝子のドットプロットとバイオリンプロットを生成するアイコン |
選択した遺伝子のクラスターの可視化
選択したマーカー遺伝子の発現分布は以下のようにして見ることができます:1)任意のマーカー遺伝子の「visualize」アイコンをクリックしてください(図 10の赤丸)。
2)選択した遺伝子のドットプロットとバイオリンプロットをそれぞれ図11aと図11bに示します。
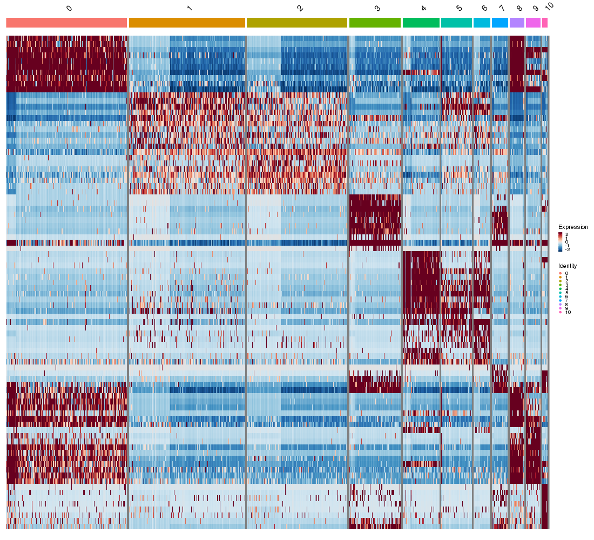
Heatmap
図12は、異なるクラスター間のマーカー遺伝子の発現をヒートマップで示したものです。
図12では、各列は一つの細胞を表しています。細胞はクラスターにグループ化されています。ヒートマップの上部にある水平の色のついたバーは、異なるクラスターを表しています。各色は特定のクラスターに対応します。
各行は同定されたマーカー遺伝子に対応します。カラースケールは遺伝子発現レベルを表します。赤は遺伝子発現が高いことを示します。青は遺伝子発現が低いことを示します。白は無発現を示します。各細胞の赤または青の強さは、各クラスター内でマーカー遺伝子がどれだけ強く発現しているかを示しています。
参考文献
1. GitHub. (2024). Single-cell RNA-seq: Marker identification. Github.
関連ブログ
- シングルセルRNA-seq解析https://basepairtech.jp/analysis/single-cell-rna-seq/
- シングルセルのマーカー遺伝子の同定https://basepairtech.jp/blog/2058/