シングルセルRNA-seqパイプラインではfastpを用いてQCとトリミングを行います。まず、UMI-toolsを用いてバーコードとユニーク分子識別子(UMI)を抽出します。次に、STARを用いてアライメントを行います。その結果、カウントマトリックスが生成されます。このカウントマトリックスはSeuratを用いたクラスタリングに使用されます。その後、マーカー遺伝子の同定とエンリッチメント解析を行うことができます。この解析が完了すると、その結果を下流の解析に利用することができます。例えば、「Single cell RNA-seq integration」パイプラインを使用して複数のサンプルの結果を比較します。
シングルセルRNA-seqパイプラインのワークフロー
図1にパイプラインのワークフローを示します。

バーコード抽出
プールされた実験中の各細胞またはRNA分子にタグ付けされたバーコードを同定し、抽出するプロセスです。
細胞バーコードは、各RNA分子の末端に付加される短い配列です。これらはライブラリーの準備中に使用されます。各細胞にはユニークなバーコードが与えられます。したがって、ある細胞からの転写産物は、全て同一のバーコードが付加されています。
細胞バーコードに加えて、ユニーク分子識別子(UMI)もシングルセルRNA-seqで使用されるバーコードの一つです。これは、各RNA分子を一意にタグ付けする短い配列です。
バーコード抽出はUMI-toolsを用いて行います。各リードの最初の数塩基が抽出され、細胞のバーコードが同定されます。次に、これらのバーコードは、実験で使用される既知のバーコードのリストと照合されます。UMIが使用されている場合は、細胞バーコードに続くリードからも抽出されます(1)。
バーコード抽出により、個々の細胞からのデータが正確に識別、分離されます。これにより、クラスタリングなどの信頼性の高いダウンストリーム解析が可能になります(1)。
品質管理(QC)とトリミング
QCはfastpを使用して実行されます。質の低いリードを評価およびフィルタリングします。シーケンス後、リード内の各ヌクレオチドに品質スコア(Qスコア)が割り当てられます。このスコアは、塩基コールに対する信憑性を示します。
トリミングは、アライメントの前にリードから不要な配列を除去するプロセスです。これにはアダプターや低品質塩基が含まれます。アダプターとは、ライブラリー調製時にRNA断片の末端に付加される短い配列のことです。これらは元のRNAの一部ではなく、シーケンスを容易にするためにのみ使用されます。
QCとトリミングにより、下流の解析に必要な高品質なデータを得ることができます(1)。
アライメント
このステップでは、STARを使用してシーケンスリードを参照ゲノムにアライメントします。それにより、シーケンスされたリードがどの遺伝子または転写産物に由来するかを決定します。
カウントマトリックスの生成
このステップでは、featureCountsを用いてリードを構造化フォーマットに変換します。構造化されたフォーマットは、個々の細胞にわたる遺伝子の発現レベルを表します。このフォーマットは「gene-cell count matrix」として知られています。行は個々の遺伝子を表します。列は個々の細胞を表します。マトリックスの各エントリーは、特定の細胞における特定の遺伝子に対応するリードの数を表します(1)。
クラスタリングと可視化
細胞はSeuratを使ってクラスターにグループ化されます。これは遺伝子発現プロファイルの類似性に基づいて行われます。したがって、各クラスターは類似した発現プロファイルを持つ細胞のグループを表します(1)。
クラスタリングの結果は3つの手法で可視化できます:
1) t-SNE (t-distributed stochastic neighbor embedding)
2) UMAP (Uniform Manifold Approximation and Projection)
3) PCA (Principal Component Analysis)
これらはすべて次元削減技術です。これらは、データを2次元または3次元空間に投影するために使用されます。これにより、同定されたクラスターを効果的に可視化することができます。したがって、異なる細胞集団にまたがる遺伝子発現の複雑なパターンの探索が容易になります(1)。
クラスターマーカー遺伝子の同定
マーカー遺伝子とは、あるクラスターにおいて他のクラスターと比較して有意な発現差を示す遺伝子のことです。これらはSeuratを用いて同定されます。これらの遺伝子はクラスターの同一性を定義するのに役立ちます。
機能アノテーション
このステップでは、同定されたマーカー遺伝子を既知の生物学的機能と結びつけます。これはGene Ontology(GO)解析とReactomeパスウェイ解析によって行われます。
Gene Ontology(GO)解析
GO解析はマーカー遺伝子のリストをインプットとして使用します。解析はマーカー遺伝子リストでどのGO用語が濃縮されているかを特定します。エンリッチメントの有意性を評価するためにp値が計算されます。これは、どの生物学的プロセス、分子機能、または細胞構成要素が、対象のマーカー遺伝子と有意に関連しているかを特定するのに役立ちます。
Reactomeパスウェイ解析
この解析では、同定されたマーカー遺伝子に有意にエンリッチされているReactomeデータベースの生物学的パスウェイを同定します。p値は濃縮の有意性を評価するために計算されます。
結果(「Report」タブ)
結果は、「Report 」タブ(図2の赤枠)にあります。
Quality scores
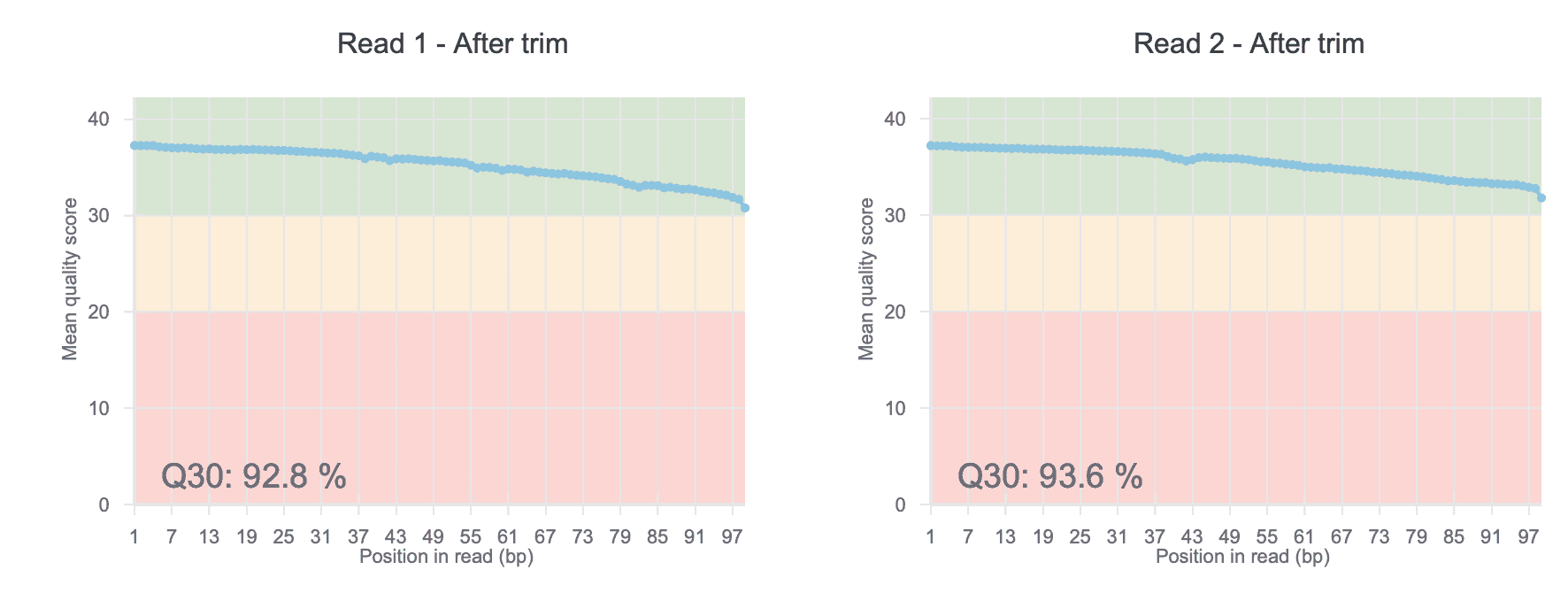
図3は、QCおよびトリミング前後の各塩基の平均品質スコア(Qスコア)を示しています。
図3では、リード1がフォワードリード、リード2がリバースリードを表します。プロットの背景は3つのQスコアセクションに分かれています。緑色の領域はQ30を表し、品質スコアが良好な領域です。オレンジ色の領域はQ20を表し、妥当なQスコアです。ピンク色の領域はQ10を表し、これは品質が悪い領域です。
図3では、すべての塩基が緑色の領域(Q30)にあります。これはリード中のすべての塩基でQスコアが良好であることを示しています。
詳細はテクニカルノート 「FASTQファイルの品質評価と前処理」をご参照ください。
Number of reads
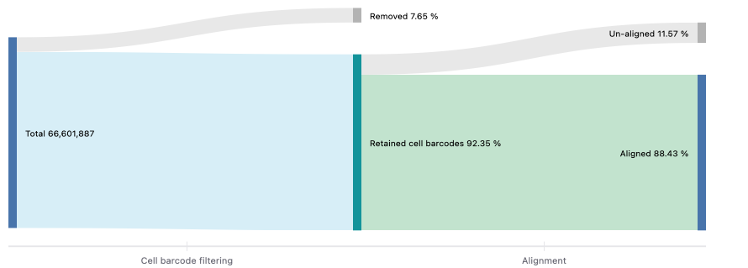
図4は、バーコードフィルタリングとアライメント前後のマッピングされたリードの割合をまとめたSankeyプロットです。
この例では、細胞バーコードフィルタリング後、約92%の細胞バーコードが保持されています。これは、これらのバーコードがフィルタリング基準に合格し、さらなる解析に有効であることを示しています。しかし、約7%のバーコードが除去されました。これは品質が低いためと考えられます。
詳細はテクニカルノート 「FASTQファイルの品質評価と前処理」をご参照ください。
Metrics
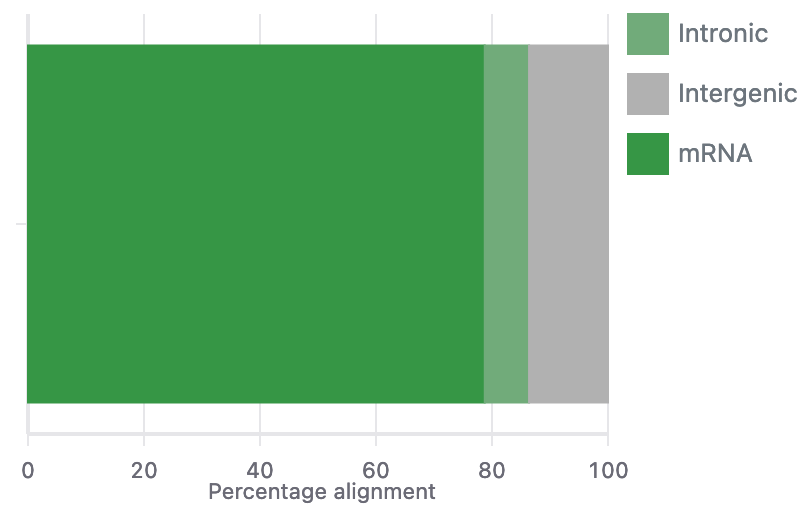
図5は、さまざまなゲノム領域でリードがどのようにアライメントされたかをまとめたプロットです。これらの領域には、mRNA、イントロン、遺伝子間領域が含まれます。
図5では、ほとんどのリードがmRNAにアライメントしています。イントロン領域と遺伝子間領域にアライメントする割合は少ないです。このアライメント分布は、一般的にシングルセルRNA-seqデータから予想されるものと一致しています。つまり、遺伝子発現を正確に測定するためには、ほとんどのリードがエクソン領域にあるべきです。
Summary
図6は、フィルタリング前後のデータに含まれる細胞と遺伝子・フィーチャーをまとめたものです。
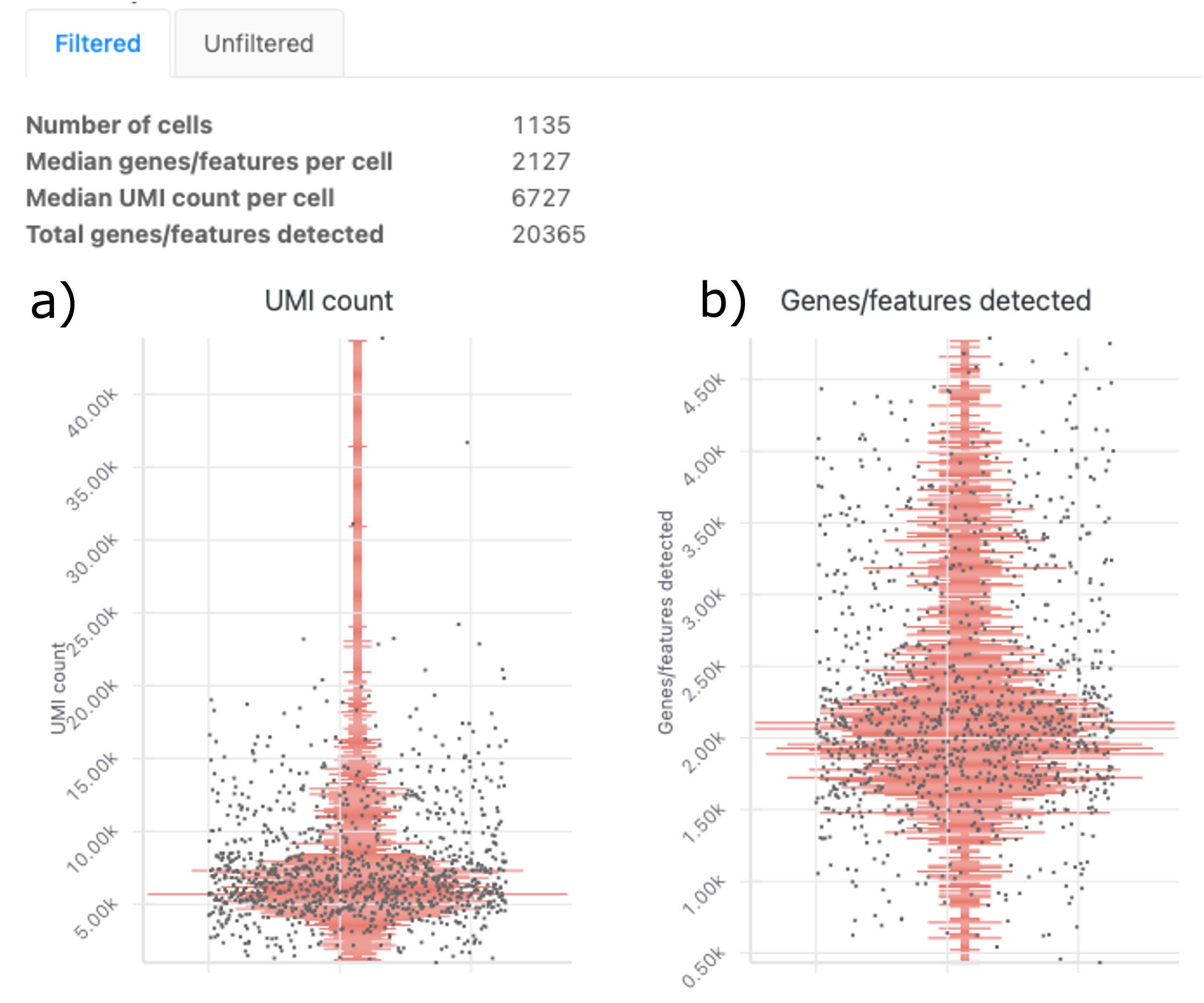
図6上段の用語
| 用語 | 定義 |
| Number of cells | 個々の細胞の総数 |
| Median genes/features per cell | 細胞あたり検出された遺伝子・フィーチャーの平均数。または、各細胞で転写産物が捕捉されたユニークな遺伝子フィーチャー数の中央値 |
| Median UMI count per cell | 細胞あたり検出された転写産物の平均数 |
| Total genes/features detected | 全細胞で検出された遺伝子・フィーチャーの総数 |
UMIカウント分布プロットを図6aに示します。各黒点は個々の細胞を表します。赤い線は、UMIカウントの一般的な密度を示しています。UMIカウントの範囲は、細胞あたり~5,000~40,000です。ほとんどの細胞のUMI数は中央値(6727 UMI/cell)前後です。
遺伝子・フィーチャー分布プロットを図6bに示します。図6aと同じように、各黒点が細胞です。赤い線は密度を表します。大半の細胞で1,500から2,500の遺伝子・フィーチャーが検出されています。中央値は細胞あたり2,127遺伝子・フィーチャーが検出されました。
Scatterplot
図7はクラスタリング中に生成されたt-SNE、UMAP、PCAプロットを示します。上部のタブ(図7青枠)をクリックすることで違うプロットを表示できます。
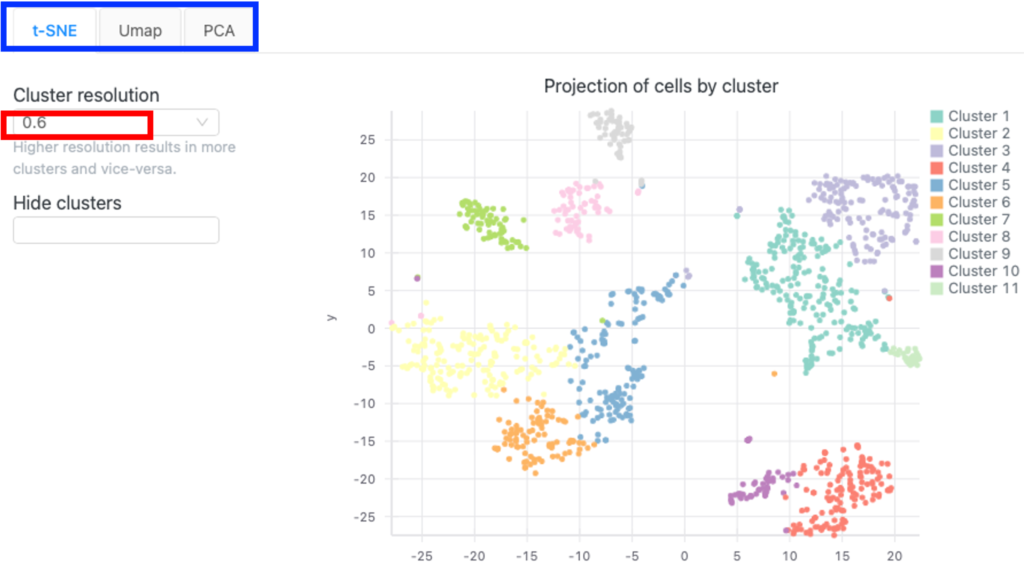
図7では、軸は高次元空間における局所的な類似性を保持する方法で、細胞の2次元投影を表しています。これらの軸の実際の単位には生物学的な解釈はありません。しかし、点間の相対的な距離には意味があります。
それぞれの点は一つの細胞を表します。このプロットで互いに近い細胞は、類似した遺伝子発現プロフィールを持ちます。したがって、それらはクラスターにグループ化できます。一方、離れている細胞はより明確な発現パターンを持ちます。
すべてのクラスターは異なる色で示されています。各クラスタ内のセルの決定はクラスタリングアルゴリズムに依存します。クラスタリングアルゴリズムはクラスタ分解能(図7の赤枠)に影響されます。クラスタ解像度は、クラスタリングアルゴリズムを適用するときに、細胞のクラスターをどの程度区別するか、あるいはどの程度細かくするかを制御するパラメータです。
図7では、細胞は分解能0.6で11のクラスターにグループ化されています。解像度の値が低いと、より少ない、より広いクラスターになります。逆に、解像度の値が高いほど、より多く、より細かいクラスターになります。
クラスターのコレスポンデンス
クラスターは、以下のものに対応します:
1) 異なる細胞タイプ
2) 異なる細胞状態:
例えば、発生の異なる段階にある細胞は、別個のクラスターを形成します。
3) 細胞タイプ内のサブタイプ:
一つの細胞型であっても、遺伝子発現の微妙な違いに基づいてクラスターを形成し、さらに細分化される可能性があります。
選択遺伝子のt-SNEプロットとバイオリンプロットの表示
図8は選択された遺伝子のa) t-SNEプロットとb) violinプロットです。図8の赤枠で遺伝子を選択すると、a) t-SNEプロットとb) violinプロットを見ることができます。これらは選択した遺伝子と分解能での発現分布を示します。
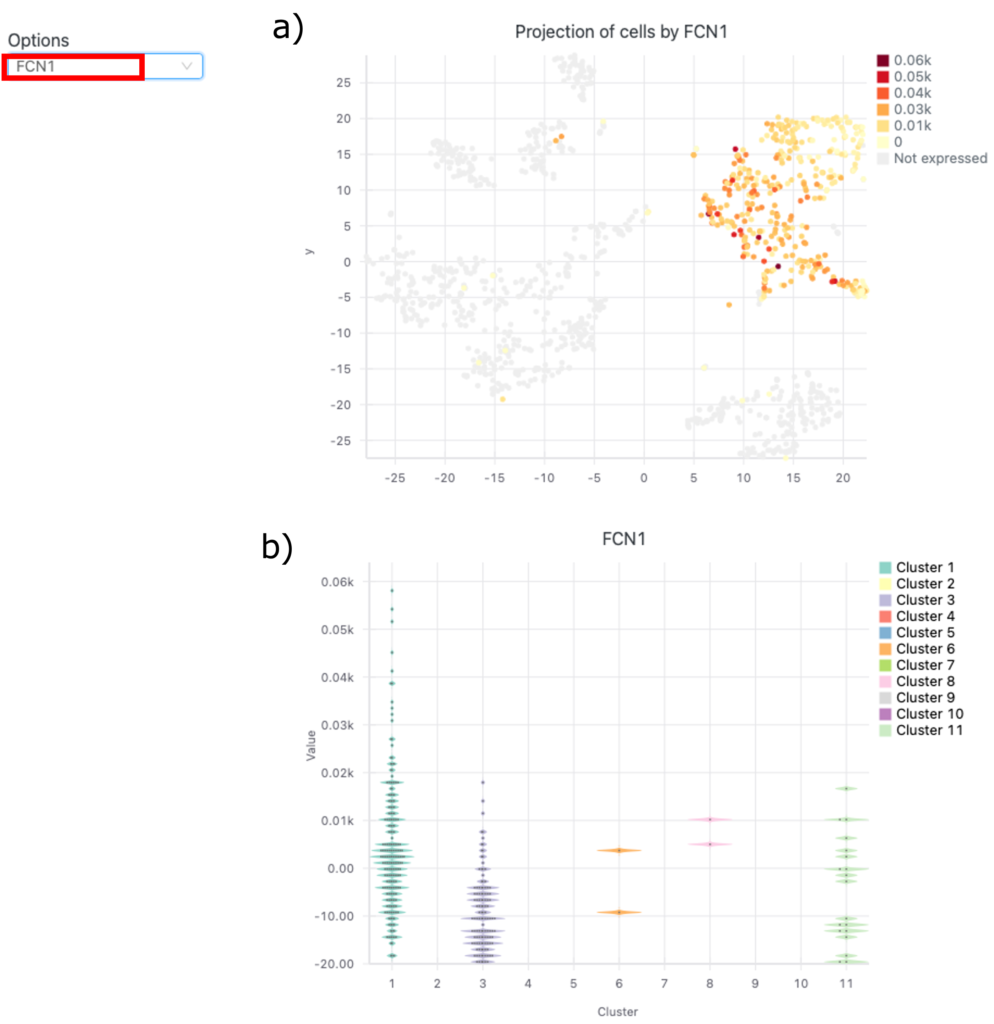
図8aでは、各点は1つの細胞を表しています。細胞の位置は遺伝子発現パターンの類似性を反映しています。カラースケールはUMIカウントを表します。各細胞における遺伝子発現レベルを測定します。UMIカウントが高い(濃い赤)ほど、転写が活発であることを示唆します。一方、カウントが低い(薄い黄色)ほど、遺伝子発現が低いことを示唆します。選択されたFCN1遺伝子の細胞の遺伝子発現はクラスター1で最も高いです。遺伝子発現レベルが低いのはクラスター3です。
図8bでは、異なる細胞クラスターにわたるFCN1遺伝子の発現が示されています。X軸は異なるクラスターを表します。Y軸は選択した遺伝子の発現レベルを示します。正の値は高い発現を表します。一方、負の値またはゼロの値は、発現が低いか全くないことを示します。バイオリンプロットの結果はt-SNEプロットの結果と一致しています(図8a)。
Cluster table
図9は、選択した分解能での各クラスタのマーカー遺伝子のリストを示します。
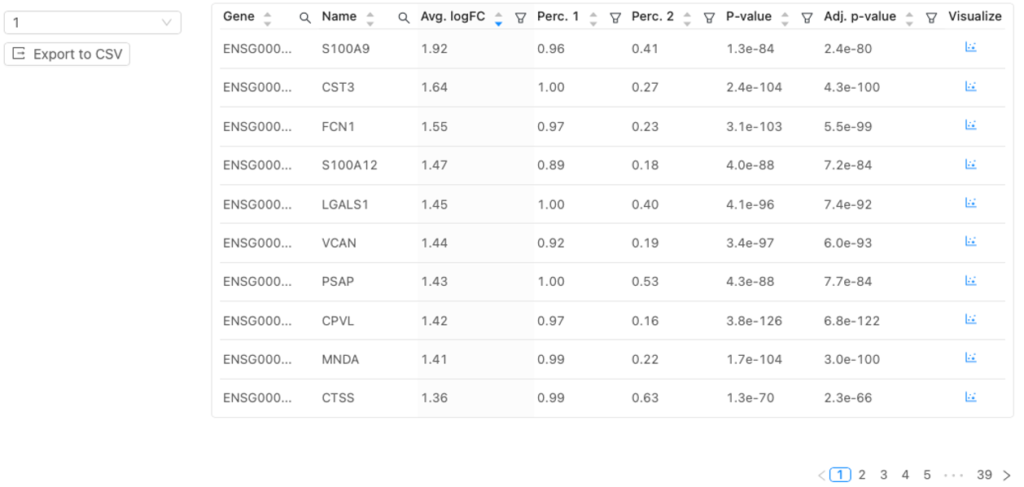
用語
| 用語 | 定義 |
| Gene | 同定されたマーカー遺伝子の記号 |
| Name | 同定されたマーカー遺伝子の名前 |
| Avg. logFC | Average log fold change。選択されたクラスターとそれ以外のクラスターにおけるこのマーカー遺伝子の遺伝子発現の差 FCが1:発現に変化なしFCが1以上:発現増加FC値<1:発現減少例えば、図9で選択されたクラスターはクラスター1です |
| Perc. 1 | 選択されたクラスターにおいて、マーカー遺伝子が発現している細胞の割合。 |
| Perc. 2 | マーカー遺伝子が発現している他の全てのクラスターにおける細胞の割合 |
| p-value | 選択されたクラスターと他のクラスターとの間の遺伝子発現の差の統計的有意性。p値が小さいほど有意差が大きいです |
| Adj. p-value | Adjusted p-value。Multiple hypothesis testingを考慮して補正されたp値。つまり、検出された偽陽性結果を補正する尺度 |
GO table
図10は、選択した分解能における各クラスターのGO用語の濃縮結果を示しています。
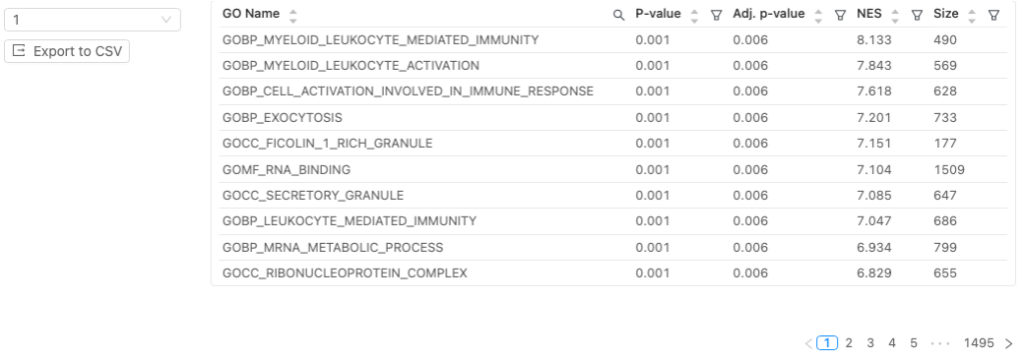
用語
| 用語 | 定義 |
| GO Name | Gene Ontology用語 |
| p-value | 特定のGOタームに関連する遺伝子の濃縮の統計的有意性 |
| Adj. p-value | 調整p値。多重仮説検定を考慮してp値に適用される補正。すなわち、検出された偽陽性の結果を補正する尺度 |
| NES | 正規化エンリッチメントスコア。遺伝子セットのサイズのばらつきに対して正規化されたエンリッチメントスコア |
| Size | GO用語に属するクラスター内の遺伝子数 |
Pathway table
図11に、選択した分解能での各クラスタのReactomeパスウェイエンリッチメントの結果を示します。
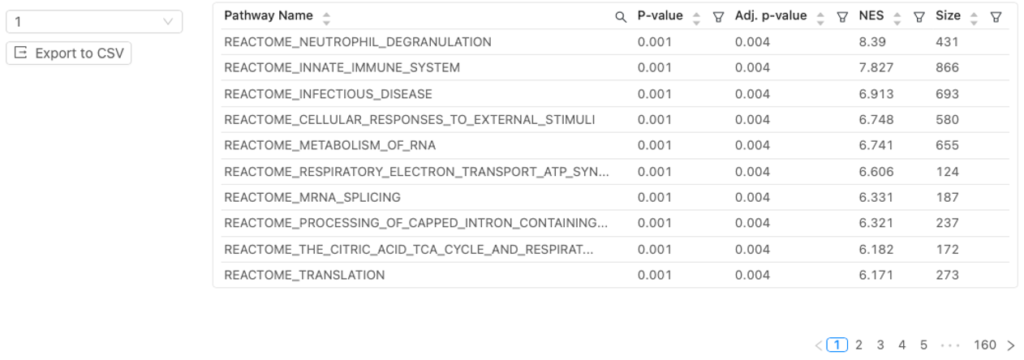
用語
| Term | 定義 |
| Pathway name | Reactomeデータベースの生物パスウェイ |
| p-value | 特定のパスウェイに関連する遺伝子のエンリッチメントの統計的有意性 |
| Adj. p-value | 調整p値。多重仮説検定を考慮してp値に適用される補正。すなわち、検出された偽陽性の結果を補正する尺度 |
| NES | 正規化エンリッチメントスコア。パスウェイのサイズのばらつきに対して正規化されたエンリッチメントスコア |
| Size | 特定のパスウェイに関与するクラスター内の遺伝子数 |
Heatmap
図12は、異なる細胞クラスターにわたるマーカー遺伝子の発現をヒートマップで示したものです。
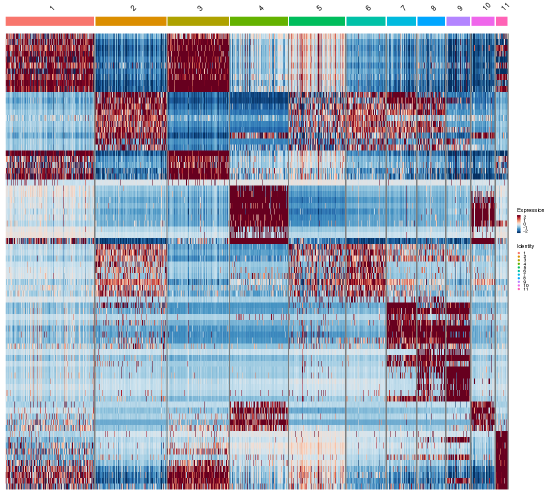
図12では、各列が細胞のクラスターを表しています。各行はそのクラスターで同定されたマーカー遺伝子に対応します。カラースケールは遺伝子発現レベルを表します。濃い赤は高発現を示します。濃い青は発現が低いか全くないことを示します。赤から青へのグラデーションは発現レベルの変化を示します。
参考文献
関連ブログ
シングルセルRNA-seq解析 https://basepairtech.jp/analysis/single-cell-rna-seq/