
DESeq2パイプラインが実行されると、最初にExpression count(STAR)パイプラインが自動的に実行されます。DESeq2パイプラインはSTARパイプラインで生成された遺伝子カウントマトリックスを使用します。まず初めに、発現変動遺伝子解析を行います。次に、PCA解析とPairwise correlationを実行します。最後に遺伝子セットのエンリッチメント解析を行います。
Basepairには2つのDESeq2パイプラインがあります:
- 「DESeq2, compare 2 groups」パイプライン:2つの異なるサンプルグループの比較に使用されます。
- 「DESeq2, 3+ group comparison」パイプライン:3つ以上の異なるサンプルグループの比較に使用されます。
パイプラインのワークフロー
図1にDESeq2パイプラインのワークフローを示します。

発現変動遺伝子解析
この解析では、DESeq2を用いて発現変動遺伝子(Differentially expressed genes、DEG)を同定します。DEGはサンプルグループ間で有意な発現変化を示す遺伝子です。DESeq2はSTARパイプラインで生成された生カウントマトリックスをインプットとして使用します。
まず、生カウントが正規化されます。生カウントは遺伝子あたりのリードカウントです。正規化は差異を考慮するために行われます。これには、サンプル間のシーケンス深度やライブラリーサイズの違いが含まれます。次に、DESeq2は各遺伝子のカウントデータをフィットするために負の二項モデルを使用します。このモデルは生物学的および技術的変動性を考慮します。
その後、各遺伝子について「log2 fold change」を計算します。これはサンプルグループ間の正規化された発現レベルの比の対数(底2)です。これはサンプルグループ間の遺伝子発現の変化の大きさを表します。log2 fold changeがポジティブの場合、その遺伝子は同じグループ内で別のグループと比較して発現がアップレギュレートしています。ネガティブの場合、その遺伝子は別のサンプルグループと比較してダウンレギュレートしています。
次に、差次的発現の有意性を評価するために統計的検定が行われます。p値は発現差の有意性を評価するために計算されます。
Principal component解析(PCA)
PCAは次元削減技術です。RNA-seqデータには多くの遺伝子が含まれているため、PCAはこの複雑なデータセットをより少ない数の線形無相関変数に削減します。これらの変数を用いて遺伝子発現データのばらつきを説明します。使用される変数はPrincipal component(PC)と呼ばれます。これにより、サンプル間の遺伝子発現のばらつきを視覚化しやすくなります。
Pairwise correlation解析
この解析はサンプル間の遺伝子の類似性を評価するために用いられます。この解析では、各サンプルペア間の相関係数を計算します。計算には、正規化された生カウントデータが使用されます。正規化された値はサンプル間の相対的な遺伝子発現を表します。これらの値を用いて一対相関を計算します。
機能アノテーション
このステップでは、同定されたDEGを既知の生物学的機能と結びつけます。これはGene Ontology(GO)解析とReactomeパスウェイ解析で行われます。
Gene Ontology(GO)解析
GO解析は、DEGsの中でエンリッチされているGO用語の同定に役立ちます。この解析では、DESeq2によって同定されたDEGのリストをインプットとして使用します。また、GOデータベースからの遺伝子セットも入力として使用します。GOデータベースの各遺伝子は、その生物学的プロセス、分子機能、細胞構成要素に関連する用語でアノテーションされています。
GO解析は、特定のGO用語に関連するDEGの数を、偶然にその用語にアノテーションされた遺伝子の予想数と比較します。統計的検定は、バックグラウンドセット(例えば、すべての発現遺伝子)と比較して、特定のGO用語がDEGのリストで有意にエンリッチされているかどうかを評価するために実行されます。その結果、DEGにおいて統計的にエンリッチされた GO用語のリストが得られます。
Reactomeパスウェイ解析
この解析では、DEGsに有意にエンリッチされた生物学的パスウェイを同定します。この解析ではDESeq2で同定されたDEGのリストをインプットとして使用します。また、Reactomeパスウェイデータベースのパスウェイをインプットとして使用します。
この解析ではDEGをReactomeパスウェイにマップします。パスウェイに関連するDEGの数が偶然発生するよりも多いかどうかを計算します。エンリッチメントの有意性を評価するために統計的検定が行われます。その結果、DEGに統計的にエンリッチされたパスウェイのリストが得られます。
結果(「Report」タブ)
すべての結果は、「Report」タブ(図2の赤枠)にあります。
Volcano plot and heatmap
図2aと図2bは、それぞれDESeq2によって生成されたボルケーノプロットとヒートマップを示しています。
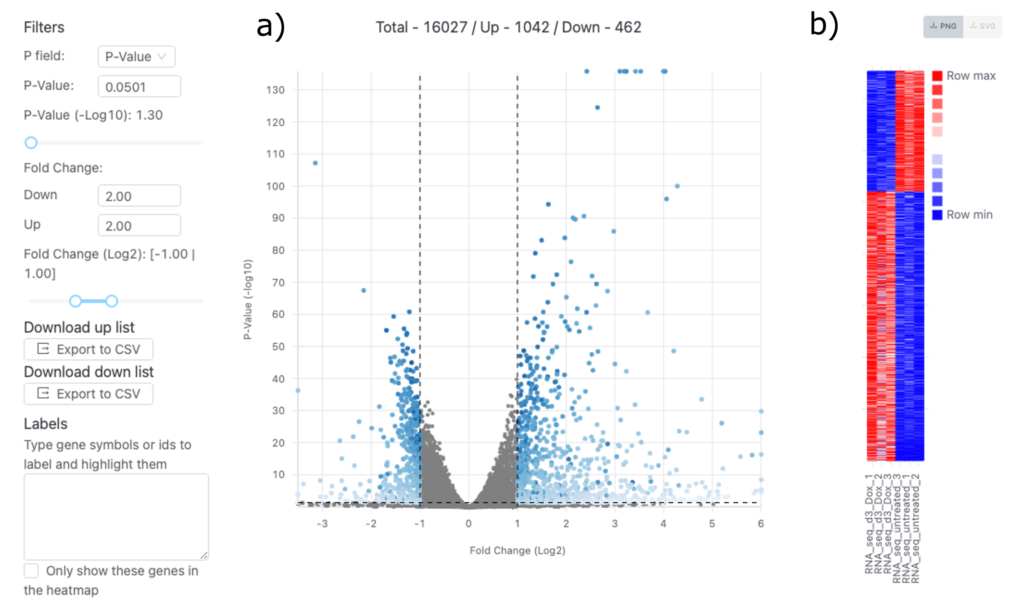
Volcano plot
X軸とy軸
図3aにおいて、x軸はサンプルグループ間の遺伝子発現のlog2倍変化を表します。0の左にある遺伝子はダウンレギュレートされています。これは負のlog2倍変化で表されます。一方、右側の遺伝子はアップレギュレートされています。これは正のlog2 fold changeで表されます。遺伝子が0から遠ければ遠いほど、fold changeは大きくなります。
Y軸はp-値の-log10を示します。すなわち、発現差の有意性を示します。y軸上で高い遺伝子ほど統計的に有意です。値が大きいほどp値が小さいことを意味します。従って、統計的有意性が高いことを示します。例えば、Y軸の20の点は10−20のp値に対応します。
破線
破線の縦線はlog2 fold changeの閾値を示します。これらの線の外側にある遺伝子は、生物学的に意味のある発現の変化があると考えられます。右側の遺伝子は発現が上昇しているます。一方、左側の遺伝子は発現が低下しています。
破線の横線は調整p値の閾値を示します。この線より上の遺伝子は統計的に有意です。
全体として
図3aでは、大きなフォールド変化と高い統計的有意性を持つ遺伝子が多く存在します。これらはプロット上に散在する青い点で示されています。アップレギュレーションを示す遺伝子の数も注目に値します。これはポジティブのlog2倍変化で示されています。また、多くの遺伝子がダウンレギュレーションを示します(ネガティブのlog2 fold change)。最も統計的に有意な遺伝子は、プロット上でより高い位置に表示されます。これは、それらの差次的発現状態に対する信頼性を示しています。
Heatmap
図3bでは、各行が遺伝子を表しています。遺伝子は発現パターンに基づいてクラスタ化されています。これは、サンプルグループ間で類似した挙動をする遺伝子グループを強調するのに役立ちます。各列はサンプルを表します。同じグループのサンプルは一緒にグループ化されます。
ヒートマップの色は、異なるサンプル間でのその遺伝子の相対的な発現レベルを示します。赤は高発現を示します。青は低発現を示します。薄い色調は中間の発現レベルを示します。
図3bのヒートマップは、未処理のサンプルと比較して、処理したサンプルは多くの遺伝子発現において有意な変化を示していることを示唆しています。
Diff expr genes
図4はDEGのリストです。これらはサンプルグループ間で発現レベルに有意差を示す遺伝子です。
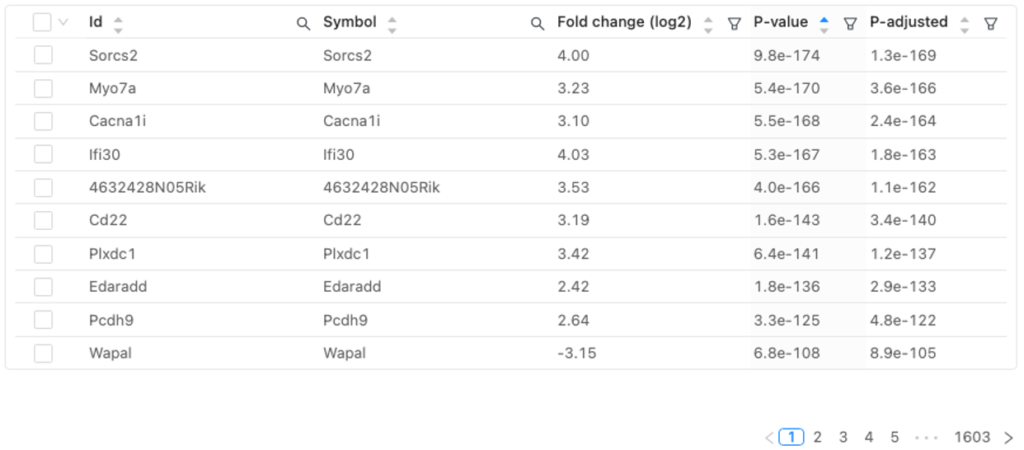
用語
| 用語 | 定義 |
| Id | 各遺伝子のユニークな識別子 |
| Symbol | 遺伝子名の略称 |
| Fold change (log2) | サンプルグループ間の発現のLog2比。正の値はその遺伝子がアップレギュレーションしていることを示します。負の値はその遺伝子の発現低下を示します |
| P-value | 各遺伝子の発現差の統計的有意性 |
| P-adjusted | 偽陽性をコントロールするために多重検定で調整したp値 |
PCA
図5にサンプルのPCA結果を示します。
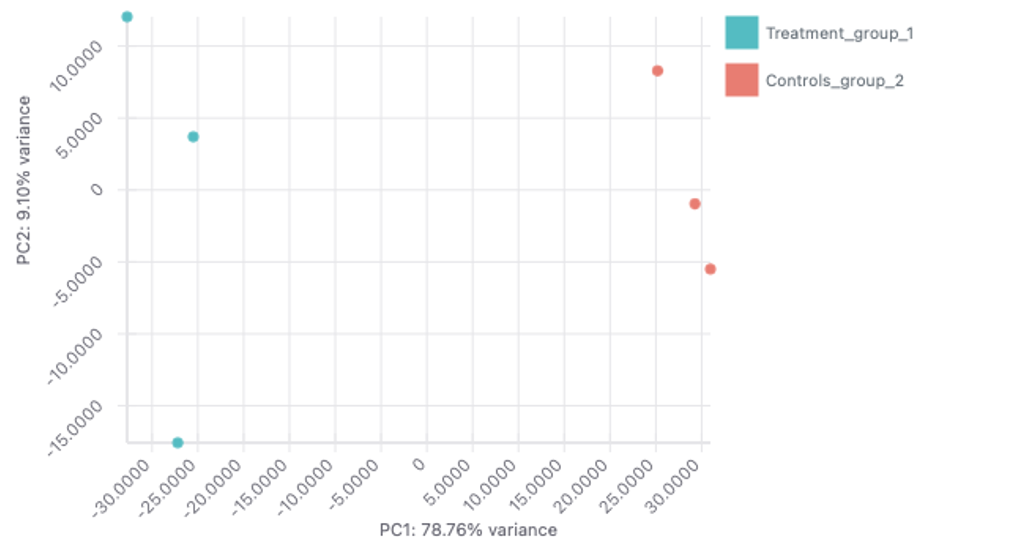
PCAは、異なるサンプルグループが遺伝子発現に基づいて明確に分かれているかどうかを明らかにするのに役立ちます。例えば、プロット上で異なるサンプルグループが明確に分離している場合、異なるサンプルグループが異なる遺伝子発現変化と関連していることを示します。PCAはまた、各グループのサンプルが互いに生物学的類似性を有するかどうかを明らかにすることができます。例えば、サンプルグループのサンプルが密接に関連している場合、それらは類似した遺伝子発現プロファイルを持っている可能性が高いことを示します。したがって、これは生物学的類似性を示しています。
図5では、x軸が主成分1(PC1)を表しています。これはデータの最も大きな分散を表しています。y軸は主成分2(PC2)を表します。これはデータ中の2番目に大きな分散を表します。各成分は互いに相関していません。各データ・ポイントは標本を表します。同じグループの標本は同じ色です。
図5では、異なるグループのサンプルの分離が有意です。これは、サンプルグループ間に強い分散があることを示唆しています。さらに、各グループのサンプル内にはクラスタリングが見られます。これは、同じグループのサンプル間の遺伝子発現プロファイルが類似していることを示しています。
Correlation plot
図6にpairwise correlationプロットを示します。
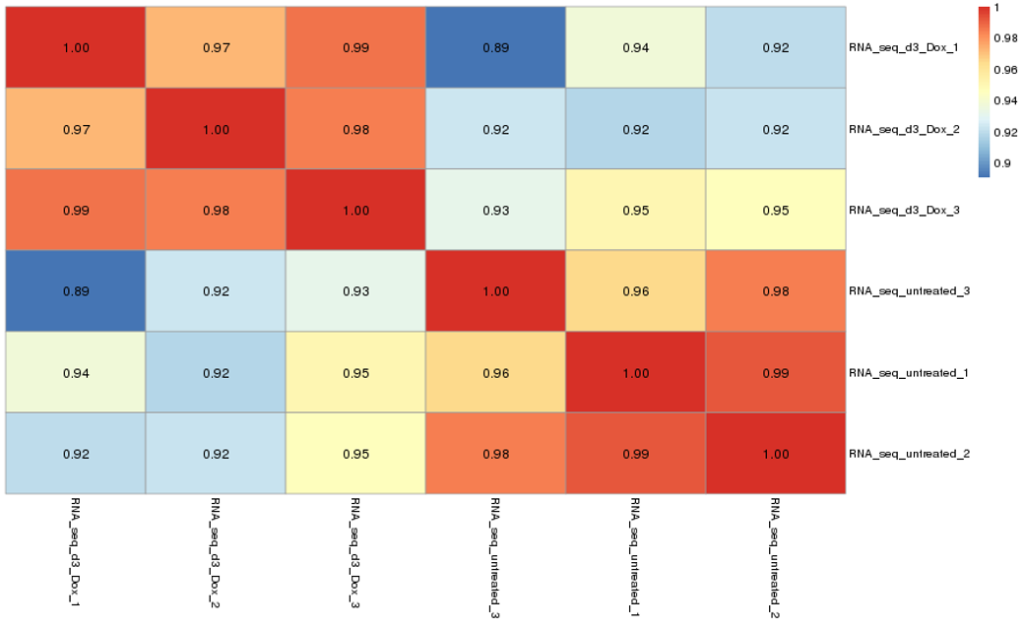
相関係数は-1~1の間の数値で、2つのサンプルの発現値がどの程度相関しているかを数値化したものです。完全な正の相関は1、相関なしは0、完全な負の相関は-1で示されます。
各セルの色の濃さは、2つのサンプル間の相関の強さを反映します。相関の高いサンプルは赤色になります。これは遺伝子発現プロファイルの類似性を示しています。一方、相関が低いサンプルは青色になります。これは遺伝子発現プロファイルの相違を示します。
GSEA GO
図7は、DEGにおいて統計的にエンリッチされたGO遺伝子セットのリストです。アップレギュレートされた遺伝子セットは「Treatment_group_1」タブの下に示されています(図7a)。ダウンレギュレートされた遺伝子セットは「Controls_group_2」タブの下に示されています(図7b)。GSEAの詳細については、テクニカルブログ 「Basepair上でエンリッチメント解析」をご覧ください。
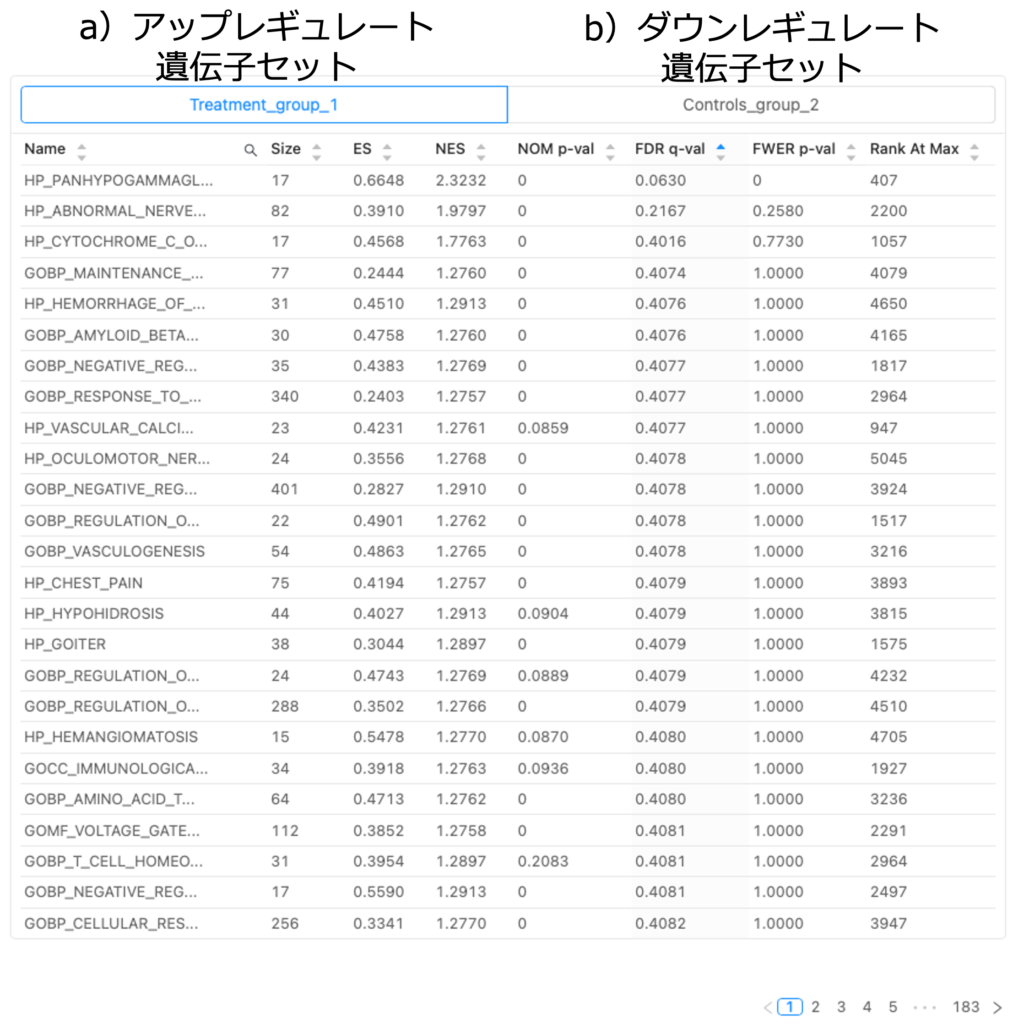
用語
| 用語 | 定義 |
| Name | GO遺伝子セット名 |
| Size | GO遺伝子セットの遺伝子数 |
| ES | エンリッチメントスコア。遺伝子セットがDEGのランク付けされたリストの一番上(アップレギュレート遺伝子)または一番下(ダウンレギュレート遺伝子)でどの程度エンリッチされているかを測定します |
| NES | Normalized enrichment score。遺伝子セットのサイズの違いを考慮して正規化されたES |
| NOM p-val | Nominal p-value。観察されたESがランダム分布と比較して有意である確率 |
| FDR q-val | False Discovery Rate q-value。多重検定コントロール後の調整p値。これは、結果が単一の偽陽性遺伝子セットを含まないことを保証します |
| FWER p-val | Family-Wise Error Rate p-value。多重検定コントロール後の、より厳密な調整済みp値。これは、結果が単一の偽陽性遺伝子セットを含まないことを保証します |
| Rank At Max | 最大ESが発生した遺伝子の順位 |
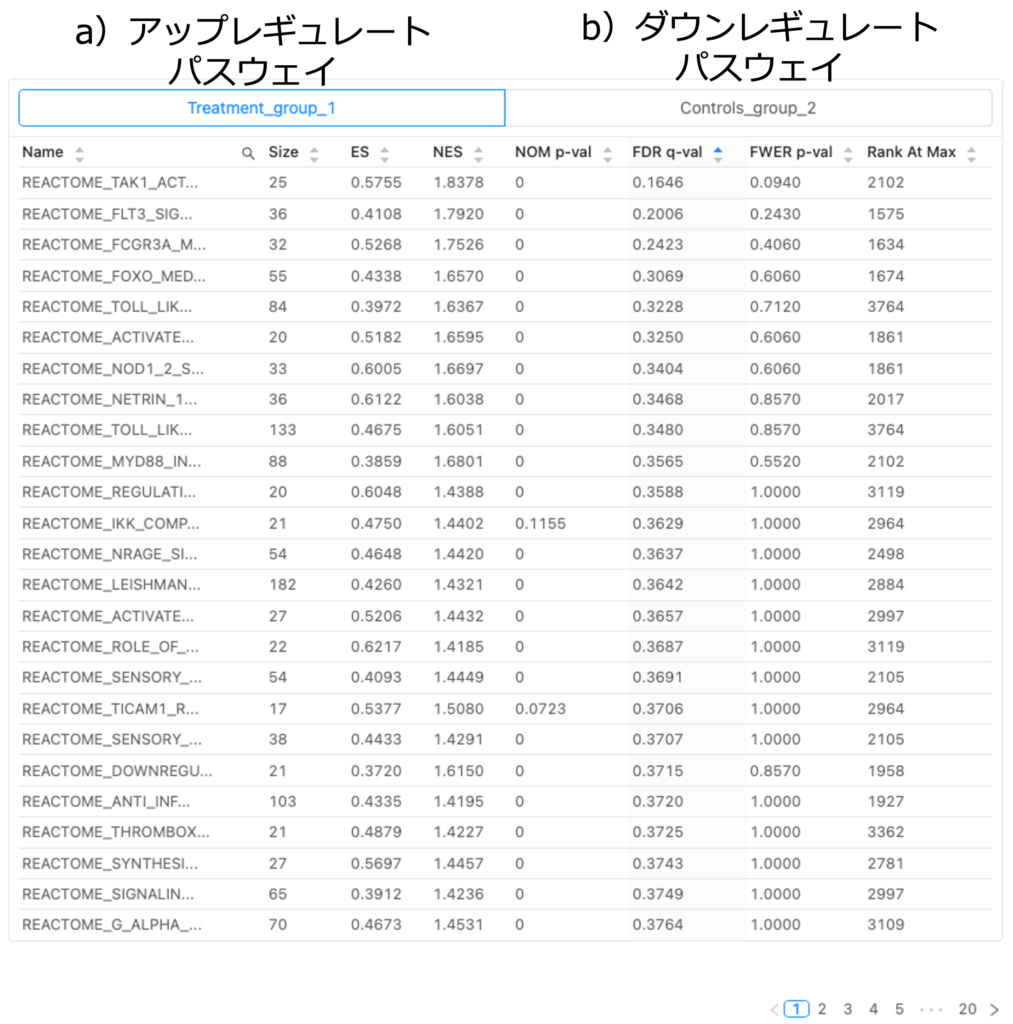
GSEA pathway
図8は、DEGにおいて統計的にエンリッチされたReactomeパスウェイのリストです。アップレギュレートされたパスウェイは「Treatment_group_1」タブに示されています(図8a)。ダウンレギュレートされたパスウェイは「Controls_group_2」タブに示されています(図8b)。図8で使用されている用語は、「GSEA GO」セクションの用語と同じです。
GSEAの詳細については、技術ブログ「Basepairでパスウェイ解析(Reactome)」をご覧ください。
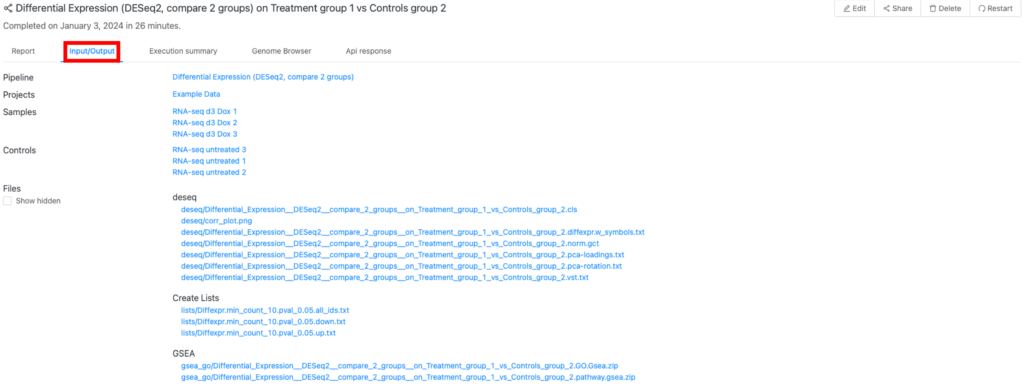
アウトプットファイル(「Input/output」タブ)
アウトプットは、「Input/output 」タブ(図9の赤枠)にあります。
DESeq
| ファイル | 説明 |
| deseq/<ANALYSIS_NAME>.cls | 発現変動遺伝子解析に使用するクラスファイル。どのサンプルがどのグループに属するかを指定するために使用します。これによりDESeq2は異なるグループ間で遺伝子発現量を比較することができます |
| deseq/corr_plot.png | 相関プロット画像ファイル。サンプル間のペア相関を可視化するために使用します |
| deseq/<ANALYSIS_NAME>.diffexpr.w_symbols.txt | 発現変動遺伝子解析結果 |
| deseq/<ANALYSIS_NAME>.norm.gct | GCT(Gene Cluster Text)フォーマットの正規化遺伝子発現マトリックスファイル |
| deseq/<ANALYSIS_NAME>.pca-loadings.txt | PCAによるprincipal componentのPCAローディング |
| deseq/<ANALYSIS_NAME>.pca-rotation.txt | PCAローテーションマトリックスファイル。サンプルがprincipal componentとどのように関連しているかの情報が含まれています |
| deseq/<ANALYSIS_NAME>.vst.txt | Variance-stabilized transformed(VST)発現データファイル。VSTはカウントデータを正規化するために適用されます。また、VSTは発現レベルの範囲にわたって分散を安定化させます。これにより、下流の解析に適したデータになります。 |
Create lists
| ファイル | 説明 |
| lists/Diffexpr.min_count_10.pval_0.05.all_ids.txt | すべての発現変動遺伝子(DEG)のリスト |
| lists/Diffexpr.min_count_10.pval_0.05.down.txt | DESeq2によるダウンレギュレート遺伝子とその結果のリスト |
| lists/Diffexpr.min_count_10.pval_0.05.up.txt | DESeq2によるアップレギュレート遺伝子とその結果のリスト |
参考文献
1. Basepair. (2020). A deep dive into differential expression. Basepair. https://www.basepairtech.com/knowledge-center/a-deep-dive-into-differential-expression/
関連ブログ
Basepair上でエンリッチメント解析 (GSEA/Gene set enrichment analysis)

Basepairでパスウェイ解析(Reactome)https://basepairtech.jp/?p=2137