
Bowtie2パイプラインはエピジェネティクスのシーケンスデータ(FASTQファイル)の前処理を行います。
パイプラインのワークフロー
以下のパイプラインはすべて同じワークフローです(図1*):
- ATAC-Seq Alignment (Bowtie2)
- ChIP-Seq QC, Alignment (Bowtie2)
- CUT&RUN QC, Alignment (Bowtie2)
- CUT&Tag QC, Alignment (Bowtie2)

図1. Alignment(Bowtie2)パイプラインのワークフロー
QC &トリミング
この最初のステップでは品質管理が行われ、fastpを用いて低品質のリードとアダプターがトリミングされます。
アライメント
このステップでは、Bowtie2を用いてリードがリファレンスゲノムにアライメントされます。
ミトコンドリアリードの除去
ミトコンドリアゲノムは(ヌクレオソームを持たないため、)Tn5酵素による切断およびアダプター挿入を受けやすい状態と考えられます。
カバレッジが高いことが多いので、samtoolsを用いてミトコンドリア由来のリードを除去します。
重複リードの除去
重複リードとは、同じゲノム領域にマップされた複数のリードのことです。これらはPCR増幅時に一部の領域が過剰増幅された結果です。重複リードはアーチファクトであり、下流解析の結果にバイアスを生じさせる可能性があるため、sambamba markdupを用いて除去します。
インサートサイズの測定
インサートサイズは、リード1とリード2のペア間の距離です。そのため、インサートサイズからリードペア由来のDNA断片のサイズが推定できます。インサートサイズはPicardを用いて測定します。DNA断片長の分布は、使用したシーケンスデータの質を評価する良い指標となります。
ゲノムカバレッジの計算
カバレッジとは、ゲノムの塩基または領域がシーケンスされた回数です。ゲノムカバレッジは、BEDToolsを使用し、リファレンスゲノム全体にアライメントされたリードのカバレッジを計算します。
結果(「Report」タブ)
上記パイプラインで生成された結果は、「Report」タブにあります。
ここでは、ATAC-seqの結果例を示します(図2の赤枠)。
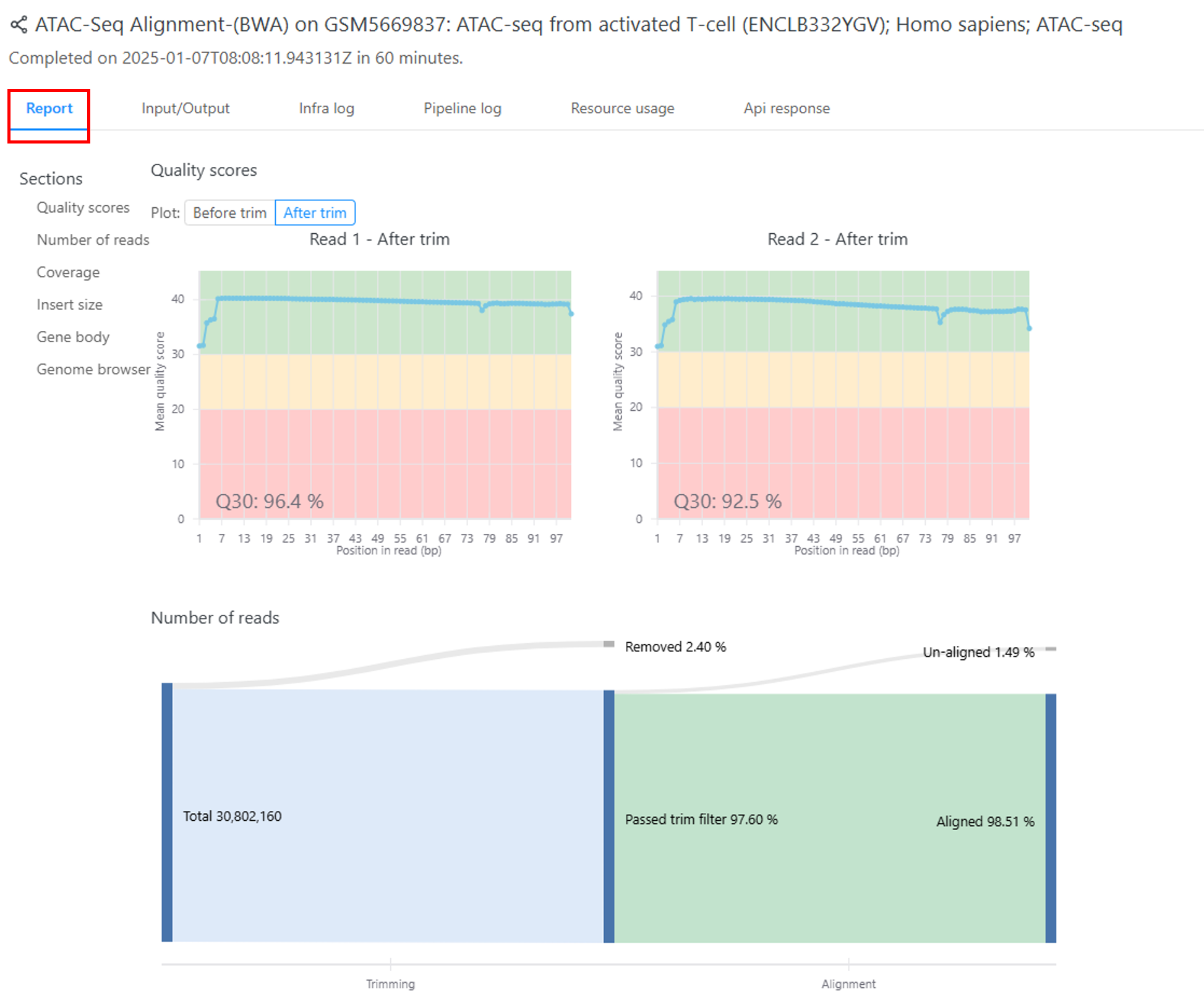
図2. 「ATAC-seq Alignment(Bowtie2)」パイプラインの結果例
Quality score
品質管理およびトリミング前後の塩基ごとの平均Q scoreを図3に示します。
詳細は「FASTQファイルの品質評価と前処理」のテクニカルノートをご覧ください。
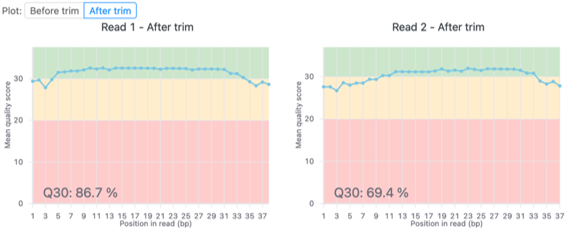
図3. リード1 (左) とリード2 (右) における、トリミング前後の塩基あたりのQ score
Number of reads
マップされたリードの割合をまとめたリバープロットを図4に示します。詳細は「FASTQファイルの品質評価と前処理」のテクニカルノートをご覧ください。
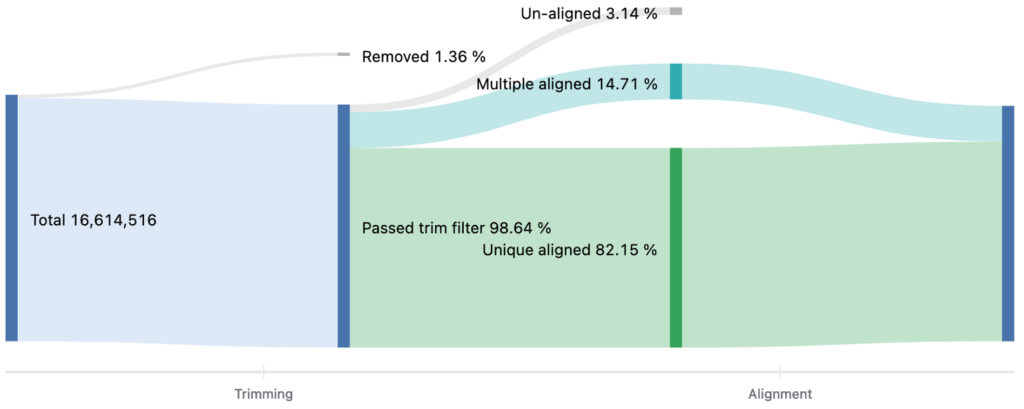
図4. マップされたリードの割合をまとめたリバープロット
Coverage
ゲノム全体のシーケンスカバレッジをサマリーしたプロットを図5に示します。ここでは、X軸は各塩基の読まれた回数であるカバレッジを表します。また、Y軸はリードがカバーする塩基の割合を表します。
図5の曲線は、低いカバレッジ値ではほぼ100%の塩基から始まり、ほとんどすべての塩基が最小限のカバレッジであることを示しています。カバレッジ値が大きくなるにつれて、塩基の割合は徐々に減少します。これは、カバレッジ値が高いほどカバーされる塩基が少なくなることを示しており、ゲノムのすべての領域に等しくアクセスできないATAC-seqデータでは想定される結果です。高いカバレッジでは、曲線は0に近い値になっていることから、ほとんどの塩基が低いカバレッジであることを示します。
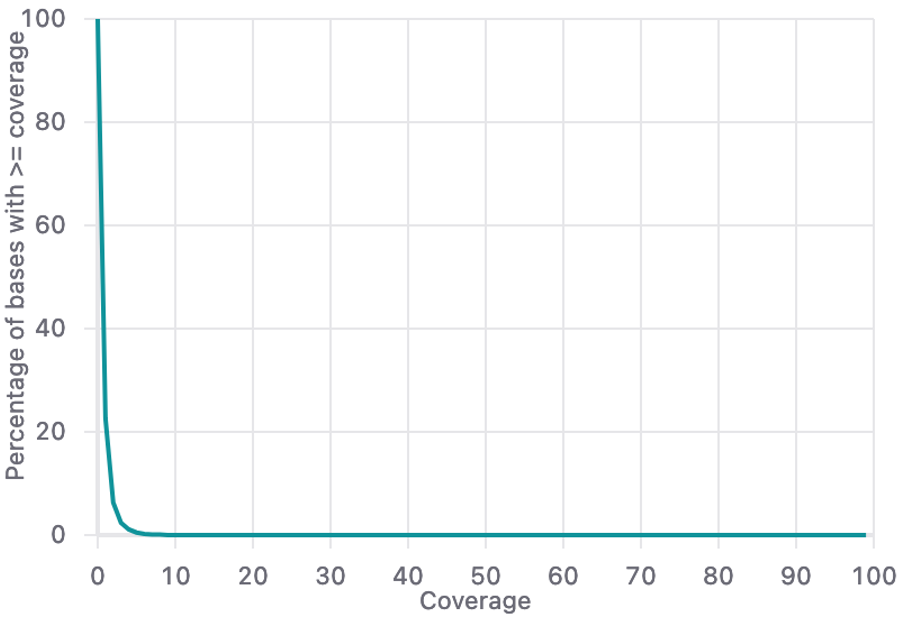
図5. リードカバレッジサマリープロット
Insert size
ペアエンドデータのインサートサイズの分布を図6に示します。ここでは、X軸はインサートサイズ(塩基対)を表し、Y軸はそのインサートサイズのリード数を表します。
図6のプロットでは、50塩基対のインサートサイズ付近に非常に高いピークを示しています。これはヌクレオソームのない領域を示しており、ヌクレオソームに巻きついていないオープンクロマチン領域の存在を示しています。つまり、これらの領域は通常、転写因子のような制御エレメントが結合する場所です。
200塩基対と400塩基対のあたりに小さなピークはそれぞれ、モノヌクレオソーム断片とジヌクレオソーム断片に該当します。このパターンはクロマチンのヌクレオソーム結合領域の存在を示しており、これらの領域はアクセスしにくいが、ATAC-seqでは検出可能であることを示唆しています。
全体的な分布は、インサートサイズがジヌクレオソームサイズを超えて大きくなるにつれて、カウント数が急激に減少しています。つまり、より大きなDNA断片があまり存在しないことを示唆しており、おそらくATAC-seqで使用されるトランスポザーゼ酵素がアクセスしにくい、より密に詰まったクロマチン領域を表しているためです。
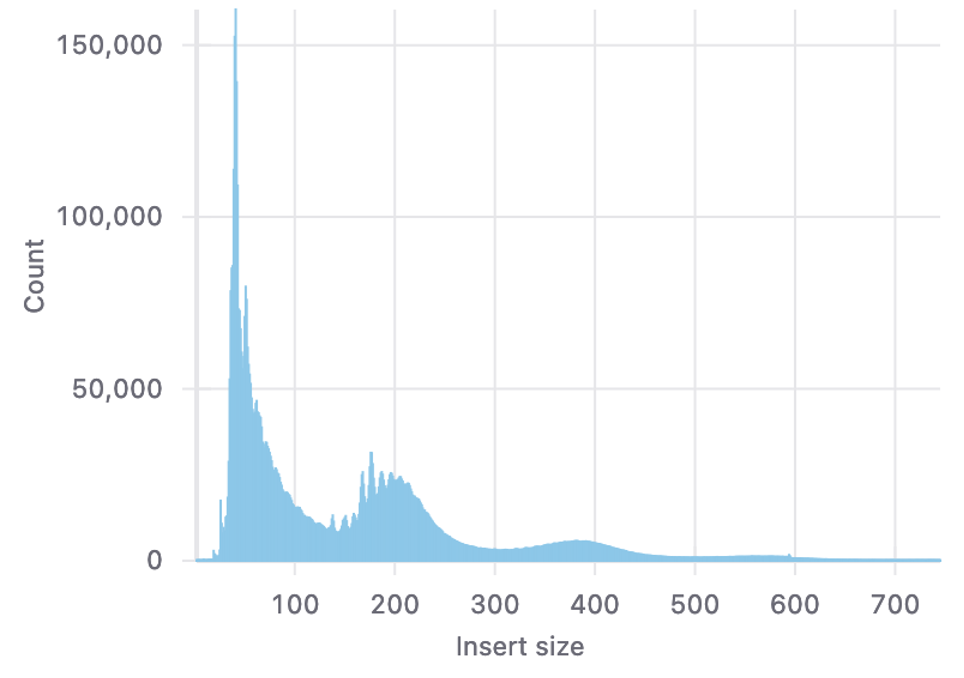
図6. インサートサイズ分布図
Gene Body and Transcription Start Site
遺伝子本体と転写開始点(Transcription start site、TSS)における正規化シグナルの平均プロファイルとヒートマップを図7に示します。
Gene Body
ATAC-seqでは、Gene bodyとは、TSSから始まり、エクソンとイントロンを経て転写終了点(Transcription end site、TES)までの領域を指します。つまり、エクソン、イントロン、非翻訳領域(Untranslated regions、UTR)もGene bodyに含まれます。
Gene bodyの平均プロファイル(図7a)では、X軸は遺伝子開始(左)から遺伝子終了(右)までの距離を示し、Y軸は正規化シグナルを示します。この例では、遺伝子開始点近傍にアクセシビリティの顕著なピークがあり、これはおそらくプロモーター領域に該当しています。さらに、シグナルはGene bodyに沿って減少しており、プロモーター領域と比較してGene body内のクロマチンアクセシビリティが低いことを示しています。
Gene bodyのヒートマップ(図7c)において、X軸はTSSからの距離(左)、Y軸は個々のGene bodyを表します。色の強度は正規化されたシグナルを表し、色が濃いほどアクセシビリティが高いことを示します。これは平均的なプロファイルと一致し、多くの遺伝子においてTSSでアクセシビリティが高く、その後Gene bodyに沿ってシグナルが減少します。
Transcription Start Site (TSS)
TSSの平均プロファイル(図7b)では、X軸がTSSからの距離、Y軸が正規化シグナルを示しています。この例では、TSSにピークがあり、プロモーターにおけるクロマチンアクセシビリティが非常に高いことを示しています。シグナルはTSSの両側で対称的に減少しており、TSSから離れるにつれてアクセシビリティが低下していることを示しています。
TSSのヒートマップ(図7d)では、X軸はTSSからの距離、Y軸は個々のTSSを表しています。これは平均的なプロファイルと一致して、多くの遺伝子でTSSでのアクセシビリティが高く、TSSの両側で対称的に減少しています。
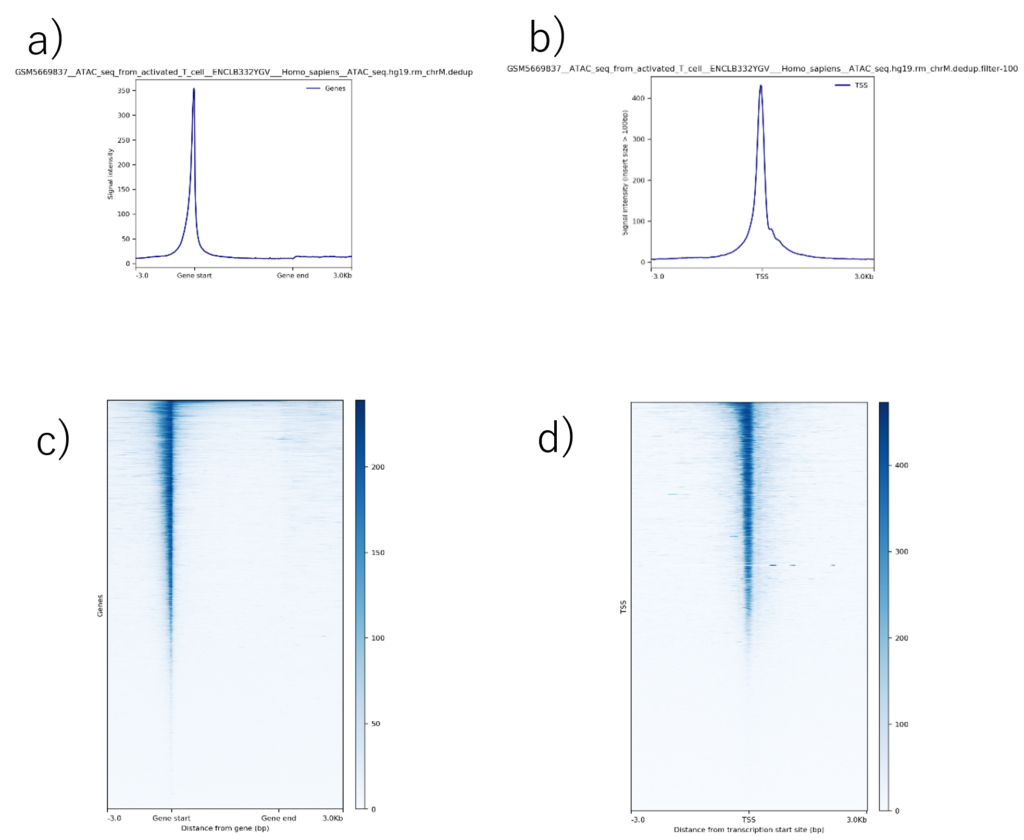
図7. a)Gene bodyとb)TSSの平均プロファイル。c)Gene bodyとd)TSSのヒートマップ
Genome browser
Integrative Genomics Viewer(IGV)でシグナル強度を視覚化した例を図8に示します。
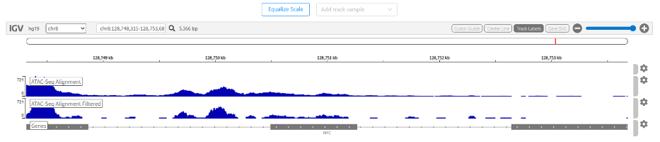
図8. IGVでシグナル強度を可視化した例
アウトプットファイル(「Input/output」タブ)
パイプラインによって生成されたすべてのアウトプットファイルは、「Input/output」タブ(赤枠)にあります。
以下はATAC-seqにおける表示例です。
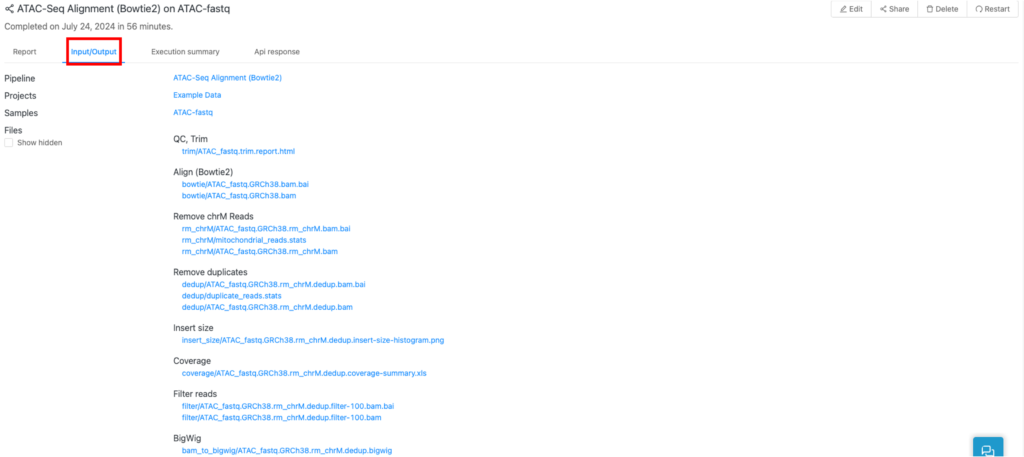
パイプラインによってアウトプットファイルの種類が異なるため、以降はファイルごとに、表示されるパイプラインを以下のマークで示します。
マーク:パイプライン名
A:ATAC-Seq Alignment (Bowtie2)
C:ChIP-Seq QC, Alignment (Bowtie2)
R:CUT&RUN QC, Alignment (Bowtie2)
T:CUT&Tag QC, Alignment (Bowtie2)
QC, Trim
- 生のシーケンスデータの品質、GC含量、PCR重複率の推定値、インサートサイズの分布、アダプター含量、kmerカウント数などの詳細なレポート(trim/<SAMPLE_NAME>.trim.report.html) [A, C, R, T]
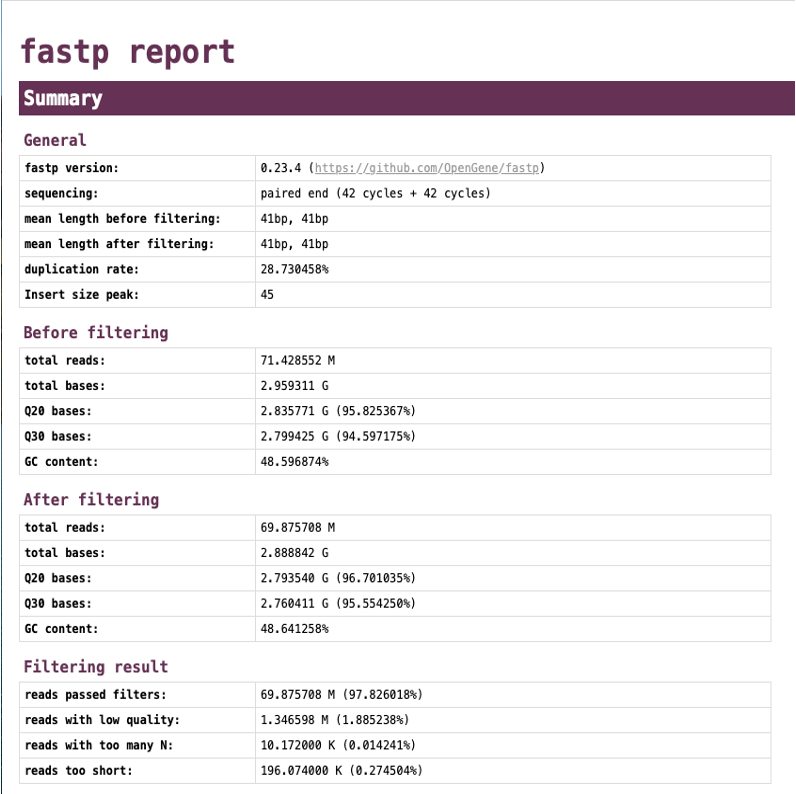
Align (Bowtie2) ※CUT&RUN、CUT&Tagの場合は”Proper Aligned”と表記
- アライメントファイルのインデックスファイル
(bowtie/<SAMPLE_NAME>.<genome>.bam.bai) [A, C, R, T] - アライメントBAMファイル(BAM形式)
(bowtie/<SAMPLE_NAME>.<genome>.bam) [A, C, R, T]
Remove chrM reads
- ミトコンドリア除去後のアラインメントファイルのインデックスファイル
(rm_chrM/<SAMPLE_NAME>.<genome>.rm_chrM.bam.bai) [A, R, T] - ミトコンドリアリード除去に関する統計データ(テキスト形式)
(rm_chrM/mitochondrial_reads.stats) [A, R, T] - ミトコンドリアリード除去後のアライメントファイル(BAM形式)
(rm_chrM/<SAMPLE_NAME>.<genome.rm_chrM.bam) [A, R, T]
Remove duplicates
- ミトコンドリアリードおよび重複リード除去後のアライメントのインデックスファイル
(dedup/duplicate_reads.stats) [A, C, R, T] - 重複リード除去に関する統計データ(テキスト形式)
(dedup/duplicate_reads.stats) [A, C, R, T] - ミトコンドリアリードおよび重複リード除去後のアライメントファイル(BAM形式)
(dedup/. .rm_chrM.dedup.bam) [A, C, R, T]
Insert size
- インサートサイズの分布(PNG形式)
(insert_size/. .rm_chrM.dedup.insert-size-histogram.png) [A, C, R, T]
Coverage
- ゲノムワイドなカバレッジの要約統計量(Excel形式)
(coverage/<SAMPLE_NAME>.<genome>.rm_chrM.dedup.coverage-summary.xls) [A, C, R, T]
Filter reads
- インサートサイズが100bp未満のアライメントファイル(BAM形式)
(filter/<SAMPLE_NAME>.<genome>.rm_chrM.dedup.filter-100.bam) [A] - インサートサイズが100bp未満のアライメントのインデックスファイル
(filter/<SAMPLE_NAME>.<genome>.rm_chrM.dedup.filter-100.bam.bai) [A]
Bigwig
- 正規化されたシグナルをIGVで表示するためのファイル(bigwig形式)
(bam_to_bigwig/. .rm_chrM.dedup.bigwig) [A, C]
(bigwig/sambamba_view/. .bigwig) [R, T]
- サブヌクレオソームフラグメント(インサートサイズ100bp未満)の正規化シグナルをIGVで表示するためのファイル(BigWig形式)
(bam_to_bigwig_filtered/<SAMPLE_NAME><genome>.rm_chrM.dedup.filter-100.bigwig) [A]
- サブヌクレオソームフラグメント(インサートサイズ0~120bp)の正規化シグナルをIGVで表示するためのファイル(BigWig形式)
(bigwig_0_120/sambamba_sort_0_120/<SAMPLE_NAME>.<genome>.0_120bp.bigwig) [R, T]
- サブヌクレオソームフラグメント(インサートサイズ120~700bp)の正規化シグナルをIGVで表示するためのファイル(BigWig形式)
(bigwig_120_700/sambamba_sort_120_700/<SAMPLE_NAME>.<genome>.120_700bp.bigwig) [R, T]
Summary
- ゲノム内の単一の位置にアライメントされたリード(Unique)、複数の位置にアライメントされたリード(Multiple)、およびアライメントされていないリード(No)の要約図(PNG形式)
(summary/<SAMPLE_NAME>.<genome>.alignment-summary.png) [C, R, T]
View
- ゲノム座標順にソートされたアライメントファイル(BAM形式)
(sambamba_view/..bam) [R, T] - ゲノム座標順にソートされたアライメントのインデックスファイル
(sambamba_view/..bam.bai) [R, T]
関連ブログ
ATAC-seq https://basepairtech.jp/analysis/atac-seq/
ChIP-seq https://basepairtech.jp/analysis/chip-seq/
Cut&Run および Cut&tag https://basepairtech.jp/analysis/cutnrun-cutntag/
FASTQファイルの品質評価と前処理 https://basepairtech.jp/blog/2102/