
De-novo assembly(Trinity)パイプラインは、リファレンスゲノムなしで全長の転写産物を再構築するために使用されます。このパイプラインでは、まず品質管理と低品質リードのトリミングを行います。次に、短いRNA-seqリードを長い配列にアセンブルします。最後に、アセンブルされた転写産物を定量します。
パイプラインのワークフロー
図1にパイプラインのワークフローを示します。

リードアセンブリー
Trinityは3つのコアステップで構成されています。Inchworm、Chrysalis、Butterflyです。これらのステップはトランスクリプトームをアセンブリーするために連動します。
Inchworm
まず、RNA-seqリードはk-merに分割されます。k-merはRNA配列に由来する長さkの短い配列です。例えば、AGCTという配列では、3-merはAGCとGCTです。次に、最も頻度の高いk-merが同定され、その後、コンティグにアセンブルされます。このステップでは、最も頻度の高い転写産物を取得します。しかし、転写産物の複雑さやアイソフォームは考慮されていません
Chrysalis
Chrysalisでは、Inchwormで得られたコンティグを、配列の類似性などに基づいてグループ化します。次に、クラスタごとにde Bruijnグラフを構築し、コンティグ間の重複部分やリードによるつなぎ目を利用して、より精密な配列構造を表します。
Butterfly
このステップでは、Chrysalisが構築したde Bruijnグラフを詳細に解析し、最も支持の高い経路を探索します。これにより、アイソフォームなど密接に関連した配列を分離しながら、全長に近い転写産物配列が生成されます。
アセンブリ品質評価
転写産物はリファレンスゲノムなしで再構築されるため、エラーが起こりやすいです。したがって、品質評価によって、アセンブリが真のトランスクリプトームを反映しているのかを評価します。これは下流の解析の信頼性を確保するために行われます。
転写物の定量化
アセンブルされた転写産物にマッピングされたリードを定量化するプロセスです。これにより、転写産物の存在量の推定値が得られます。
結果(「Report」タブ)
結果は「Report」タブ(図2の赤枠)にあります。
Summary
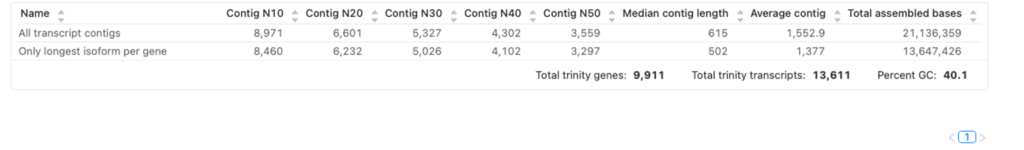
アセンブリ品質評価の結果を図3に示します。
用語
| 用語 | 定義 |
| Name | 指標の名前 |
| All transcript contigs | Trinityによって生成された全ての転写産物のコンティグを用いて計算された指標。これには選択的スプライシングイベントで生じたアイソフォームが含まれます |
| Only longest isoform per gene | 遺伝子の最長のアイソフォームのみを用いて計算された指標 |
| Contig N10, N20, N30, N40, N50 | 全アセンブリ配列長のうち、長い配列から順に合計していき、全体の10%(N10)、20%(N20)、30%(N30)、40%(N40)、50%(N50)に到達するところに位置するコンティグ長。N値が小さいほど、アセンブルの大部分に寄与しているコンティグが短いことを示します |
| Median contig length | コンティグ長の分布の中央値 |
| Average contig | アセンブルされた全コンティグの平均の長さ |
| Total assembled bases | アセンブルされた全コンティグの総塩基数 |
| Total trinity genes | Trinityによって再構築されたユニークな遺伝子の総数 |
| Total trinity transcripts | Trinityによって再構成された個々の転写産物アイソフォームの総数 |
| Percent GC | アセンブルされた全コンティグ中のguanine(G)またはcytosine(C)の塩基の割合 |
主要指標
以下は、アセンブリの品質とアウトプットを示す主要な指標です:
- Contig N50
- MedianとAverage contig length
- GC content
- Total assembled bases
- Total trinity genesとtranscripts
Contig N50
この指標はトランスクリプトームアセンブリの連続性を測定します。
図3では、2つのN50値が報告されています:
- N50 for All Transcripts
- N50 for Longest Isoform Per Gene
N50 for All Transcripts
この指標はトランスクリプトームアセンブリ全体の連続性を測定します。この指標は全遺伝子の全アイソフォームを考慮します。「N50 for All Transcript」は、全アセンブリ配列長のうち、長い配列から順に合計して50%に到達するところに位置するコンティグ長のことです。
N50値が高いほど、アセンブルされた転写産物が長く、より連続していることを示唆します。これは完全長の転写産物の再構成が良好であることを示します。一方、N50値が低いほどアセンブリが断片化していることを示します。これは、質の低いリードとアセンブル中の技術的エラーなどから生じる可能性があります。
図3では、「N50 for All Transcripts」の値は3,559です。この値は、アセンブルされた全ての転写産物の中で、アセンブリ全長の少なくとも50%をカバーするグループ内の最も短い転写産物が3,559塩基長であることを表しています。
N50 for Longest Isoform Per Gene
この指標はアセンブリの連続性を測定します。この指標は代替のアイソフォームを除外して冗長性を減らすことに重点を置いています。さらに、1遺伝子につき1つの代表的な転写産物のみを考慮します。これは遺伝子レベルでのアセンブリの質を評価するのに役立ちます。
遺伝子ごとの最長アイソフォームのN50が高いということは、アセンブリが過剰な断片化なしに個々の遺伝子の長い転写産物をうまく再構築していることを示唆します。
このN50と 「all transcripts」のN50の差が大きい場合は、代替アイソフォームの数が多いか、アセンブリに冗長性があることを示している可能性があります。
図3では、このN50値は3,297です。これは、全長の50%をカバーするグループの中で、最も短い代表的な転写産物が3,297塩基長であることを示しています。
MedianとAverage contig length
これらの指標は、転写産物の再構成の完全性を評価するために使用されます。
「Median Contig Length」は、コンティグ長の分布の中央値です。「Average Contig Length」は、アセンブリ中の全コンティグの平均長です。
中央値または平均値が高いほど 、アセンブリに長く連続した配列があることを示唆します。これらはアセンブリの質が高いことを示します。一方、長さが短い場合は、アセンブリが断片化しているか、リードの質が低いことを示します。
GC Content
これは、ゲノム中のguanine(G)とcytosine(C)塩基の割合のことです。これは、アセンブリがゲノムの予想されるGC組成と一致しているかどうかを評価するための指標です。図3では、使用したゲノムはhg19で、その典型的なGC含量は約40%です。図3では、GC含量は41%と計算されました(図3)。したがって、このアセンブリはhg19の予想されるGC含量と一致しています。
Total Assembled Bases
この指標は、アセンブリが研究対象の生物のトランスクリプトームを十分にカバーしているかどうかを決定します。「Total assembled bases」は、アセンブルされた全ての転写産物の長さの合計で、トランスクリプトームアセンブリの全長を示します。大きな値の場合はトランスクリプトームの再構築が良好で、シーケンス深度が深いことを示唆します。低い値の場合は不完全なアセンブリを示唆します。値が大きすぎる場合は、冗長性やアセンブルエラーなどの問題が発生している可能性があります。
Total trinity genesとtranscripts
これらの指標は、アセンブリによって得られたトランスクリプトームの複雑さを反映しています。
「Number of transcripts」は再構築されたアイソフォームの総数です。これは各遺伝子の全てのアイソフォームを含みます。転写産物数が多いことは、特にオルタナティブスプライシングを持つ生物において、包括的なアセンブルを示唆しています。
「Number of Genes」は、遺伝子ごとの最長のアイソフォームに基づく、遺伝子のユニークカウントの合計です。遺伝子数が多い場合は、ユニークな遺伝子をよく表現していることを示します。
「Number of transcripts」と「Number of Genes」の差が大きい場合「Number of transcripts」と「Number of Genes」の差が大きい場合は、オルタナティブスプライシングが多く、過剰な冗長性、あるいはミスアセンブリが発生している可能性があります。。
アウトプットファイル(「Input/output 」タブ)
アウトプットは、「Input/output」タブ(図4の赤枠)にあります。
Trinity
| ファイル | 説明 |
| trinity/<SAMPLE_NAME>.trinity-assembled-transcripts.fasta | アセンブルされた転写産物(fasta形式) |
| trinity/<SAMPLE_NAME>.trinity-stats.txt | アセンブルされたコンティグの統計情報の要約を含むテキストファイル |
Assembly quality assessment
| ファイル | 説明 |
| assembly_quality_assessment/<SAMPLE_NAME> .trinity.bam.bai | BAMファイルのインデックスファイル |
| assembly_quality_assessment/ <SAMPLE_NAME>.trinity.bam | アセンブルされたコンティグにアライメントされたリードのBAMファイル |
| assembly_quality_assessment/busco.zip | ゲノムアセンブリのBUSCOスコアを含むZipファイル |
Trinity transcript quantification
| ファイル | 説明 |
| transcript_quantification/ rsem_<SAMPLE_NAME>/quant_genes.results | 遺伝子ごとのリードカウントを含むタブ区切りファイル |
| transcript_quantification/ rsem_<SAMPLE_NAME>/quant_isoforms.results | アイソフォームごとのリードカウントを含むタブ区切りファイル |
参考文献
関連ブログ
BasepairのRNA-Seq https://basepairtech.jp/analysis/rna-seq/