
このパイプラインではDNA-seqのFASTQデータの前処理を行います。まず、品質評価と低品質データのトリミング、そしてアライメントを行います。次に重複リードを除去します。最後にゲノムカバレッジを計算します。
パイプラインのワークフロー
図1にパイプラインのワークフローを示します。
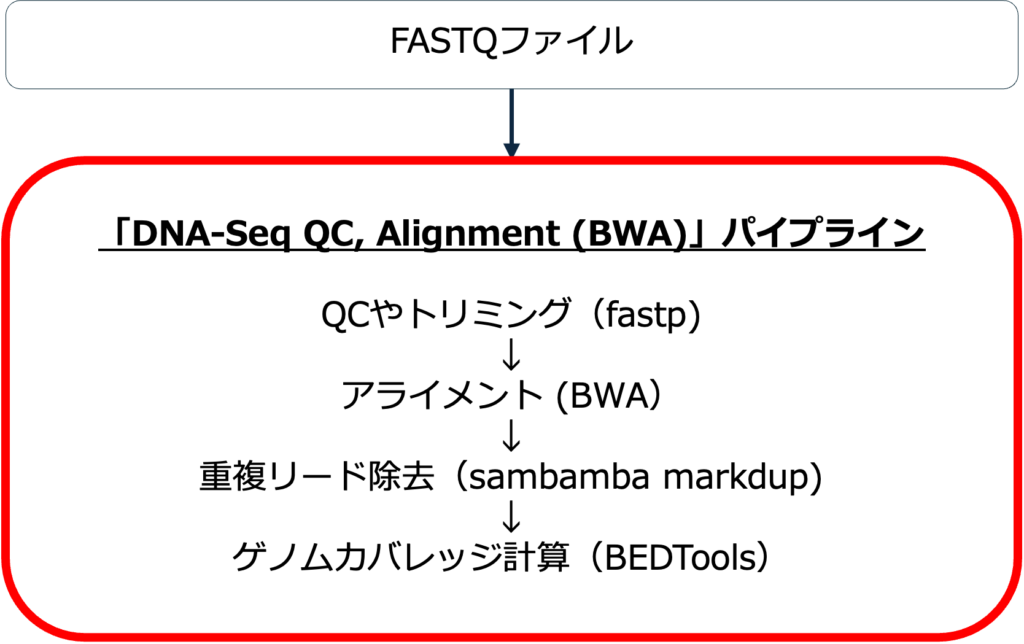
品質管理(QC) &トリミング
リードの品質はfastpを用いて評価されます。このステップでは、低品質のリードとアダプターはトリミングされます。これにより、下流の解析に適した高品質のリードが得られます。
アライメント
アライメントはBWAを用いて行います。このステップでは、トリミングされたリードをリファレンスゲノムにマッピングし、ゲノムの位置を特定します。
重複リードの除去
重複リードとは、同じゲノム領域にマップされた異なるリードのことです。これらはライブラリー調製やシーケンスプロセス中の技術的アーチファクトに起因します。例えば、PCR中の過剰増幅、シーケンス深度が深いなどです。重複リードはユニークなDNA断片ではないので、Sambama markdupによって除去されます。
ゲノムカバレッジの計算
ゲノムカバレッジとは、リファレンスゲノムの各塩基にアライメントするシーケンスリードの平均数を指し、BEDToolsを使用して計算されます。例えば、ゲノム全体に十分なシーケンス深度があるかどうかを評価するのに役立ちます。さらに、ゲノムのカバーが不十分または不均一な領域を特定するのにも役立ちます。
結果(「Report」タブ)
図2では結果を含む 「Report」タブが示されています。
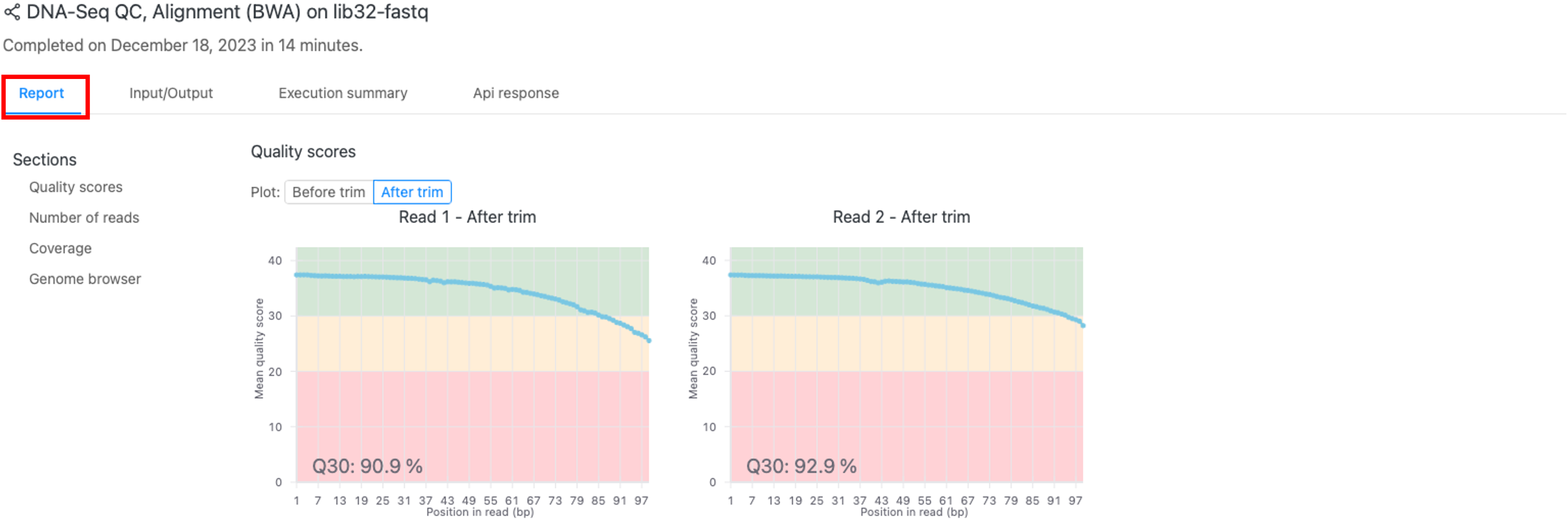
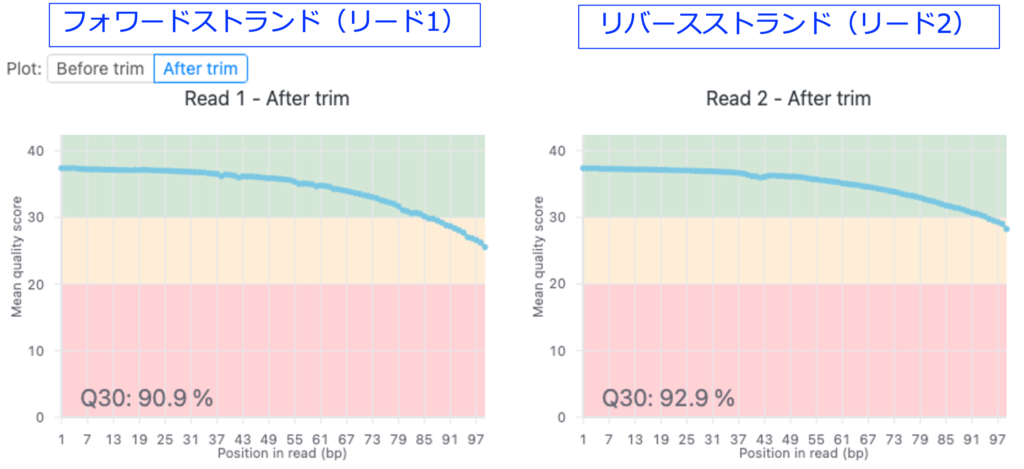
Quality scores
図3では品質管理およびトリミング前後の塩基ごとの平均のQ scoreを示します。詳細は「FASTQファイルの品質評価と前処理」のテクニカルノートをご覧ください。
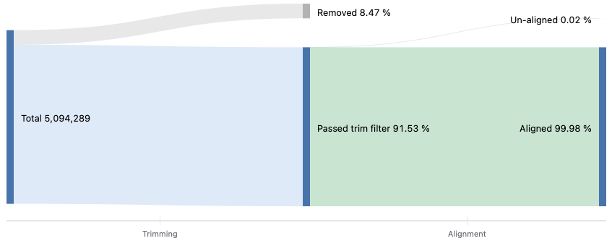
Number of reads
図4ではトリミングとアライメントの前後でマップされたリードの割合をまとめたSankeyプロットを示します。詳細は「FASTQファイルの品質評価と前処理」のテクニカルノートをご覧ください。
Coverage
図5ではカバレッジ分布図を示します。
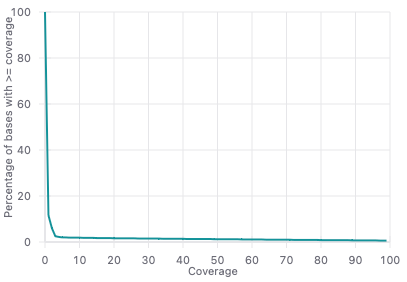
X軸はカバレッジを表します。これはゲノム中の特定の塩基位置をカバーするリードの数です。Y軸は、X軸の対応するカバレッジと同等以上のカバレッジを持つ塩基の割合(%)を表します。
図5の曲線は、低いカバレッジ値ではほぼ100%の塩基から始まり、ほとんどすべての塩基が最小限のカバレッジを持っていることを示しています。カバレッジ値が大きくなるにつれて、塩基の割合は徐々に減少します。これは、高いカバレッジを持つ塩基が少なくなっていることを示しています。このような結果はDNA-seq実験では典型的で、ゲノムのごく一部が過剰にカバーされている可能性があります。これは反復領域やGC含量などのバイアスに起因する可能性があります。
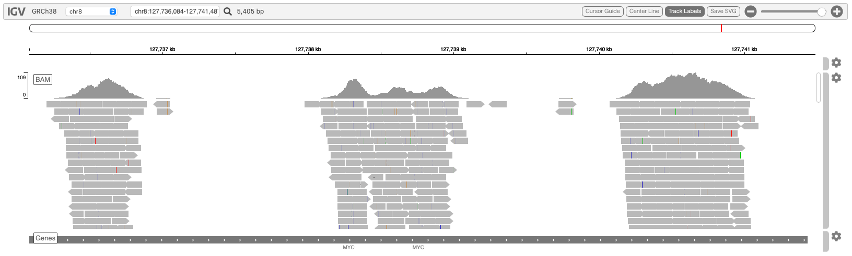
Genome browser
図6は、Integrative Genomics Viewer(IVG)に表示された正規化シグナルbigiwigトラックです。
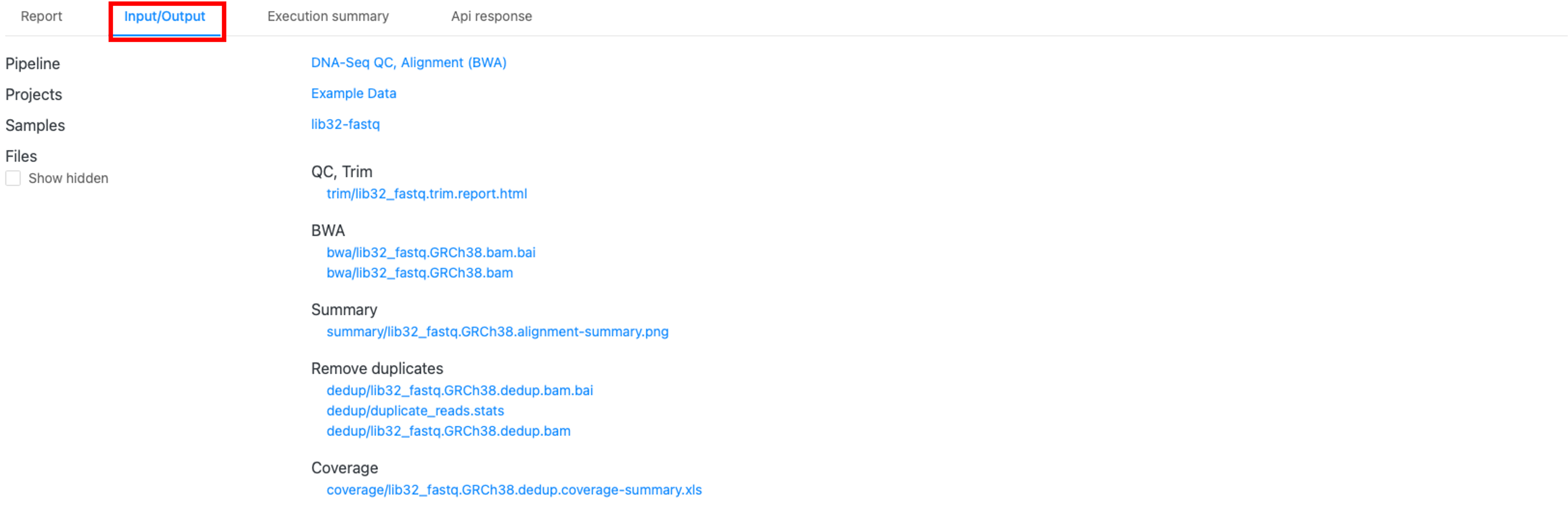
アウトプットファイル(「Input/output」タブ)
図7では、アウトプットファイルを含む 「Input/output」タブ(赤枠)が表示されています。
QC, Trim
| ファイル | 説明 |
| trim/<SAMPLE_NAME>.trim.report.html | 詳細な品質レポート(html形式) |
BWA
| ファイル | 説明 |
| bwa/<SAMPLE_NAME>.<genome>.bam.bai | アライメントbamファイルのインデックスファイル。ゲノムブラウザなどのツールで、bamファイルへの効率的なアクセスを可能にします。 |
| bwa/<SAMPLE_NAME>.<genome>.bam | 生のアライメントファイル(bam形式)アライメント |
Summary
| ファイル | 説明 |
| summary/<SAMPLE_NAME>.<genome>.alignment-summary.png | アライメントのサマリーを可視化したファイル(png形式) |
Remove duplicates
| ファイル | 説明 |
| dedup/<SAMPLE_NAME>.<genome>.dedup.bam.bai | アライメント重複リードが除去されたbamファイルのインデックスファイル |
| dedup/duplicate_reads.stats | 重複リードが除去されたアライメントの統計情報をまとめたファイル(テキスト形式) |
| leveldedup/<SAMPLE_NAME>.<genome>.dedup.bam | アライメント重複リードが除去されたアライメントファイル(bam形式) |
Coverage
| ファイル | 説明 |
| coverage/<SAMPLE_NAME>.<genome>.dedup.coverage-summary.xls | 重複が除去されたリードのゲノムカバレッジの統計情報をまとめたファイル(Excel形式) |
参考文献
1. Basepair. (2024). A comprehensive DNA sequencing analysis tutorial. Basepair. https://www.basepairtech.com/blog/dna-sequencing-analysis-tutorial/
関連ブログ
BasepairのDNA-seq https://basepairtech.jp/analysis/dna-seq-whole-genome-exome/
FASTQファイルの品質評価と前処理 https://basepairtech.jp/blog/2102/