
アウトプットファイルは、FASTQファイルの品質評価と前処理から生成されます。これらのアウトプットファイルは、各解析の「Input/output」タブ内にあり(図1の赤枠)、特に論文発表や下流の解析に役立ちます。
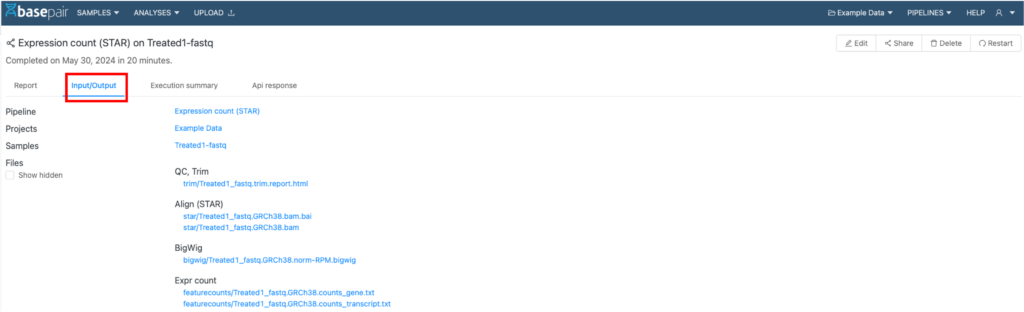
図1. 解析例の「Input/output」タブ(赤枠)
アウトプットファイル
Expression Count (STAR)パイプラインによって生成されるアウトプットファイルは以下の通りです:
QC, Trim
- 品質評価のレポート(html形式)
(trim/<SAMPLE_NAME>.trim.report.html)
Align (STAR)
- bamファイルのインデックスファイル
(star/<SAMPLE_NAME>.<genome>.bam.bai)
- アライメント生のアライメントファイル(bam形式)
(star/<SAMPLE_NAME>.<genome>.bam)
BigWig
- 正規化されたシグナルをIGVで可視化するためのファイル(bigwig形式)(bigwig/<SAMPLE_NAME>.<genome>.norm-RPM.bigwig)
Expr count
- 各遺伝子のカウントと正規化されたカウント(タブ区切りテキスト形式)
(featurecounts/<SAMPLE_NAME>.<genome>.counts_gene.txt)
- 各転写産物のカウントと正規化されたカウント(タブ区切りテキスト形式)
(featurecounts/<SAMPLE_NAME>.<genome>.counts_transcript.txt)
fastp report
fastpレポートには、シーケンスデータの品質や塩基含有量などの情報が含まれています。レポートには5つのセクションがあります:
- Summary
- Adapters
- Insert estimation
- Before filtering
- After filtering
Summary
fastpレポートの「Summary」セクションを図2に示します。
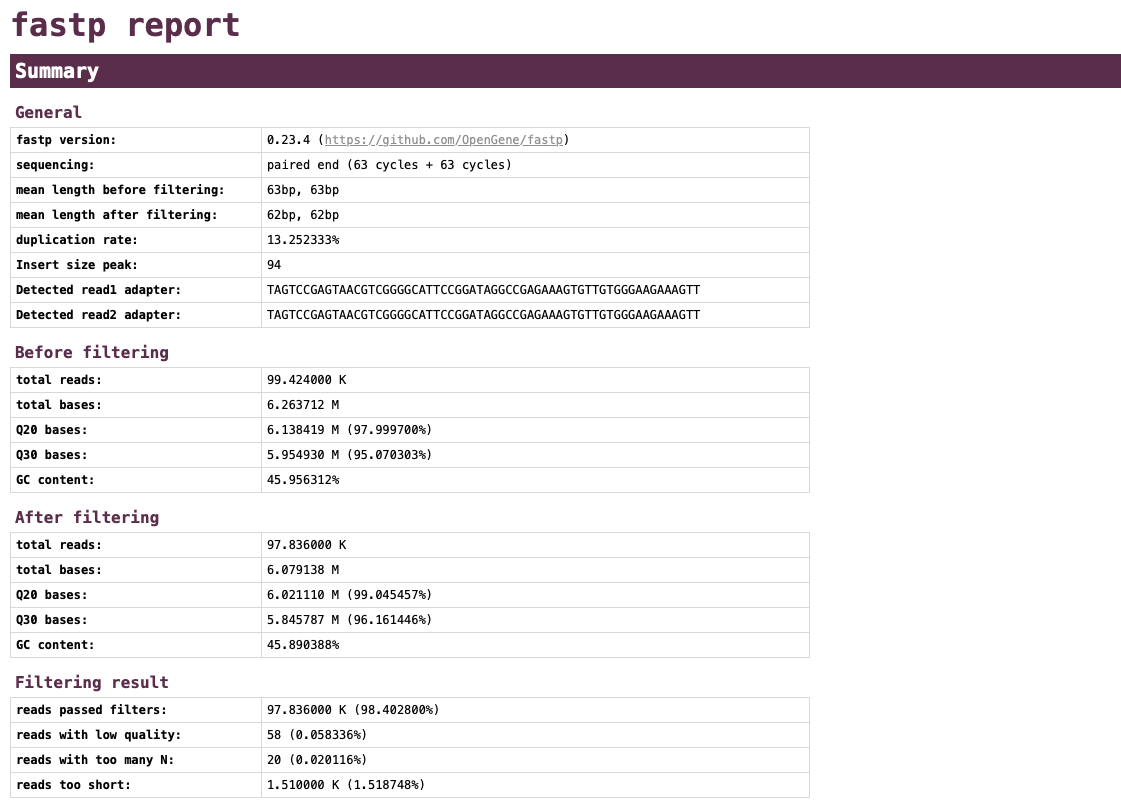
図2. fastpレポートの「Summary」セクション
「General」用語
| 用語 | 定義 |
| Fastp version | 使用したfastpのバージョン |
| Sequencing | シーケンシングのタイプ(ペアエンドなど)とサイクル数 |
| Mean length before filtering | フィルタリング前のリードの平均長 |
| Mean length after filtering | フィルタリング後のリードの平均長 |
| Duplication rate | 重複リードの割合 |
| Insert size peak | 最も多いインサートサイズ。インサートサイズとは、配列決定されたDNA断片のサイズのことです |
| Detected read1/read2 adapter | 前処理段階で検出され、トリミングされたアダプター配列 |
「Before filtering」と「After filtering」用語
| 用語 | 定義 |
| Total reads | 総リード数 |
| Total bases | 塩基配列の総数 |
| Q20 bases | Q score20以上の塩基数(およびそれらの割合) |
| Q30 bases | Q score30以上の塩基数(およびそれらの割合) |
| GC content | データ中のG塩基とC塩基の割合 |
「Filtering result」用語
| 用語 | 定義 |
| Reads passed filter | フィルターを通過したリード数と割合 |
| Reads with low quality | 低品質のためフィルタリングされたリード数と割合 |
| Reads with too many N | あいまいな塩基 (N) が多すぎるためフィルタリングされたリード数と割合 |
| Reads too short | アダプタートリミング後で、短すぎるためフィルタリングされたリード数と割合 |
Adapters
fastpレポートの「Adapters」セクションを図3に示します。このセクションには、リード1(フォワードリード)とリード2(リバースリード)の、ライブラリー調製時におけるライゲーションの不良の結果として同定された配列またはアダプター配列がリストアップされています。
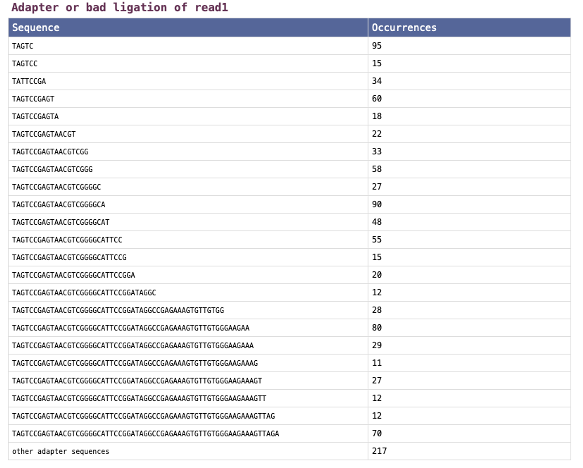
図3. fastpレポートの「Adapters」セクション
用語
| 用語 | 定義 |
| Sequences | アダプター配列として同定された配列、または不十分なライゲーションの結果として同定された配列 |
| Occurrences | 各配列がデータから検出された回数 |
| Other adapter sequences | 個別にリストアップされていないその他の配列の数 |
Insert size estimation
インサートサイズは、アダプター間で配列決定されたDNA断片の長さです。ここでは、全リードにおけるインサートサイズの分布を図4に示します。
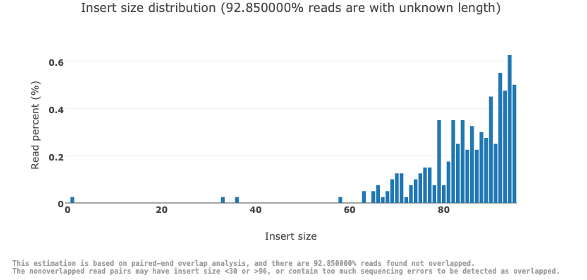
図4. 全リードにわたるインサートサイズの分布
図4では、X軸がインサートサイズ(塩基対)を表しているのに対し、Y軸は特定のインサートサイズを持つリードの割合を示します。この例では、判明しているリードの大半はインサートサイズが60から100 bpほどですが、90%以上のリードは長さが不明です。これはリードが重なっていないために、インサートサイズを直接測定できない場合に起こります。
Before and after filtering
「Before filtering」と「After filtering」には、3つのサブセクションがあります:
- Quality
- Base contents
- KMER counting
Quality
フィルタリング前のリード全体の塩基品質スコアを図5に示します。
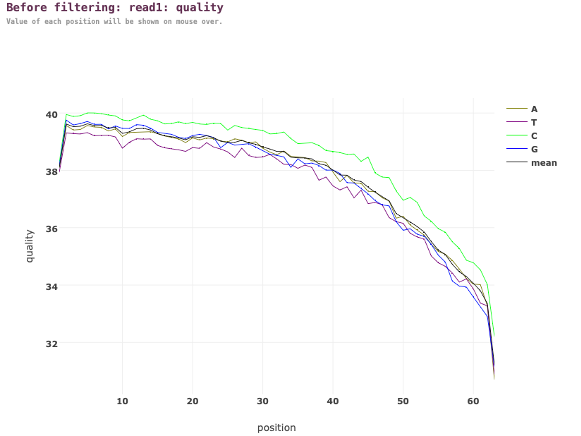
図5. フィルタリング前のリード1(フォワードリード)の塩基品質のPhredスコア
図5では、X軸は各リード内の位置を表します。一方、Y軸は各位置の塩基品質のPhredスコアを表します。Phredスコアは塩基コーリングの正確さの尺度です。したがって、Phredスコアが高いほど品質が高いです。各色の線は、各位置のすべてのリードにわたる特定のヌクレオチド(A、T、C、G)の品質スコアに対応します。
品質スコアは、リードの先頭付近(1―10位)で最も高いです。その後、リードの末尾に近づくにつれてスコアは徐々に低下します。この傾向はシーケンスデータにおいて典型的です。
Base contents
フィルタリング前のリード全体の塩基含量比を図6に示します。
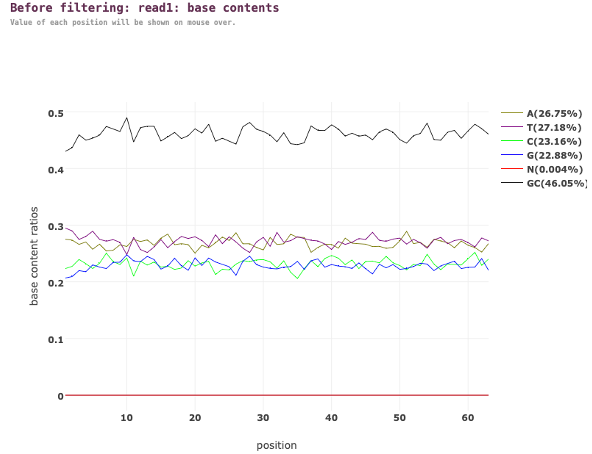
図6. フィルター処理前のリード1(フォワードリード)の塩基含量比
図6では、X軸は各リード内の位置を表します。それに対し、Y軸はリード内の各位置における各塩基の比率を表します。各色の線は、各位置における特定の塩基(A、T、C、G)の比率を表します。この例では、A、T、C、Gの比率はどの位置でも比較的安定しています。つまり、ヌクレオチドの構成がリードの長さを通して一貫していることを示唆しています。
KMER counting
K-merは、長さ「k」の短い塩基配列です。この例では、「k」はリード内のすべての5-mer(5ヌクレオチドの配列)の数を表しています。したがって、K-mer解析は、過剰に発現する配列の特定に役立ちます。つまり、特定のk-merのカウントが高い場合は、アダプターの混入、PCRアーチファクト、低品質領域を示している可能性があります。さらに、フィルタリング前後のkmer分布を比較することで、不要な配列を除去するフィルタリングステップの有効性を評価することができます。
フィルタリング前のリード1(フォワード鎖)のk-merカウントを図7に示します。

図7. フィルター処理前のリード1(フォワード鎖)のk-merのカウント
図7では、行と列がk-mer配列の1番目と2番目の位置に該当しています。各セルにはk-merが含まれます。セルの背景の濃さは、配列データ中の各k-merの相対的な存在量を示しています。例えば、セルの背景が暗いほど、リード中のk-merのカウントが高いです。
関連ブログ
FASTQファイルの品質評価と前処理 https://basepairtech.jp/blog/2102/
RNA-seqデータのトリミングとアライメントのウェビナー https://www.youtube.com/watch?v=48kHCxGMfj0&t=130s