
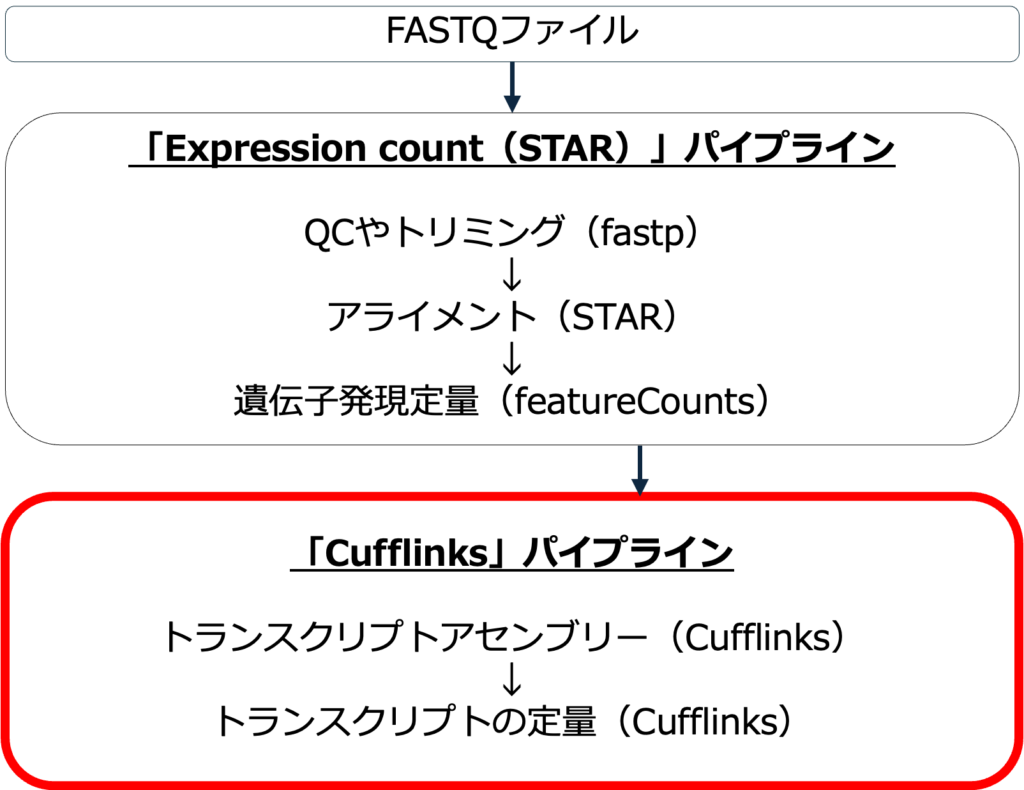
Cufflinksパイプラインを実行すると、最初にExpression count (STAR)パイプラインが自動的に実行されます。このCufflinksパイプラインは、STARパイプラインでアラインメントされたリードを使用します。まず、Cufflinksパイプラインは、リファレンスゲノムにアライメントされたリードを転写産物のセットにアセンブルします。次に、それぞれの転写産物の発現レベルを定量します。
パイプラインのワークフロー
図1にCufflinksパイプラインのワークフローを示します。
トランスクリプトアセンブリー
Cufflinksは、STARパイプラインでアラインメントされたリードをインプットとして使用します。次に、Cufflinksはアラインメントされたリードを転写産物にアセンブルします。
転写産物の定量
各転写産物について、CufflinksはFPKM (Fragments Per Kilobase of transcript per Million mapped reads)値に基づいて発現量を推定します。FPKMは遺伝子の長さとマップされたリードの総数の両方を正規化します。これは各転写産物の発現レベルを表します。したがって、サンプル間の転写産物量の比較が可能になります。
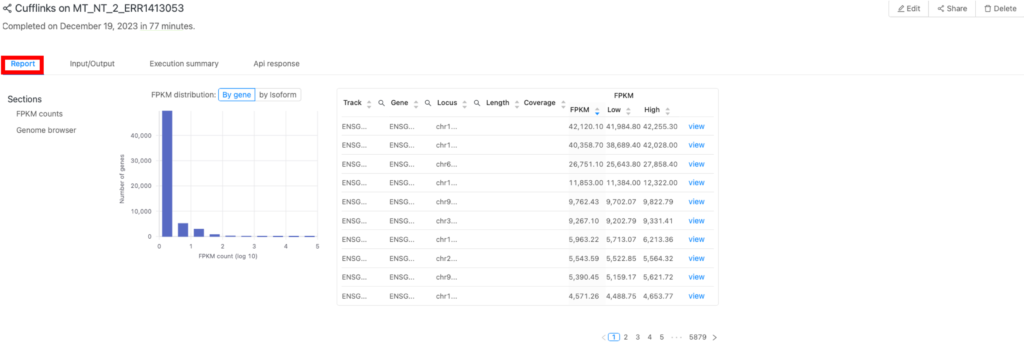
結果(「Report」タブ)
結果は「Report」タブ(図2の赤枠)にあります。
FPKM counts
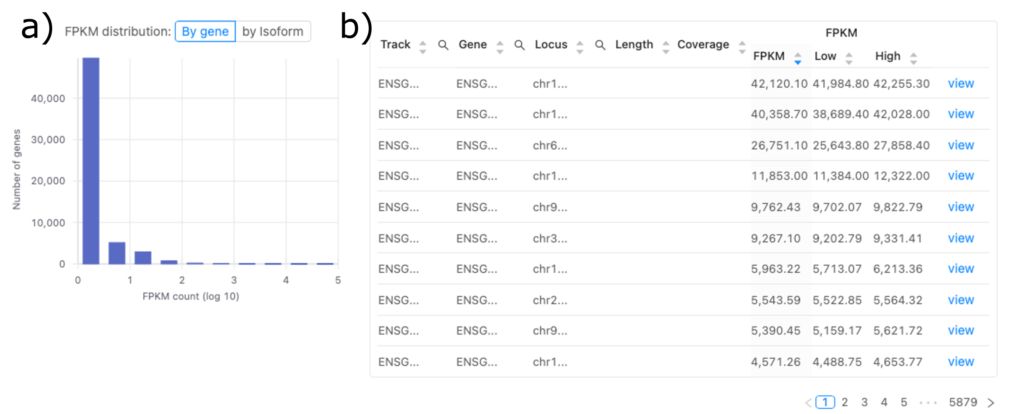
図3aでは、すべての転写産物のFPKMの頻度分布を示します。FPKM値は遺伝子発現を正規化した指標です。図3bでは、転写産物のリストとそのFPKM値を示します。
FPKMの頻度分布
図3aでは、x軸に遺伝子のFPKM値を対数スケールで表し、y軸に各FPKM値の範囲に入る遺伝子の数を表します。このデータセットでは、多くの遺伝子のFPKM値が0に近いです。これは、ほとんどの遺伝子の発現が低いか、まったくないことを示唆しています。一方、発現レベルが高い(FPKM値が高い)遺伝子は少ないです。このパターンはRNA-seqデータで想定される結果です。
遺伝子発現値のリスト
用語
| 用語 | 定義 |
| Track | 遺伝子のユニークな識別子 |
| Gene | 遺伝子の名前 |
| Locus | 遺伝子の染色体上の位置 |
| Length | 遺伝子の長さ。これはFPKMの正規化に使用されます |
| Coverage | 遺伝子のシーケンスカバレッジ |
| FPKM | Fragments Per Kilobase of transcript per Million mapped reads。遺伝子の発現量を正規化するための指標 |
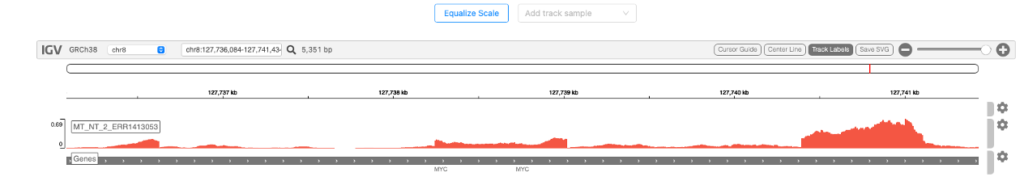
Genome browser
図4はインタラクティブなゲノムブラウザです。Integrative Genomics Viewer (IGV)でシグナル強度を視覚化できます。
アウトプットファイル(「Input/output」タブ)
図5では、アウトプットファイルを含む「Input/output」タブ(赤枠)が表示されています。

Cufflinks
| ファイル | 説明 |
| cufflinks/<SAMPLE_NAME>.<genome>.genes.fpkm_tracking | 遺伝子ごとの発現量カウントを含むfpkm_trackingファイル |
| cufflinks//<SAMPLE_NAME>.<genome>.isoforms.fpkm_tracking | アイソフォームごとの発現量カウントを含むfpkm_trackingファイル |
| cufflinks/<SAMPLE_NAME>.<genome>.skipped.gtf | Gene Transfer Format(GTF)ファイル。アセンブル時にスキップされた転写産物のリストを含みます |
| cufflinks//<SAMPLE_NAME>.<genome>.transcripts.gtf | Gene Transfer Format(GTF)ファイル。アセンブルされた転写産物のアノテーションを含みます |
参考文献
1. Basepair. (2024). RNA-seq analysis & visualization platform for both bench scientists and bioinformaticians. Basepair. https://www.basepairtech.com/analysis/rna-seq/
関連ブログ
BasepairのRNA-Seq https://basepairtech.jp/analysis/rna-seq/