
背景
RNA-seqやマイクロアレイデータのパスウェイエンリッチメント解析に最もよく使われるツールの一つは、Broad InstituteによるGene Set Enrichment Analysis(GSEA)ツールです[1,2]。GSEAで最もよく使用されるパスウェイのデータベースは、同じくBroad InstituteによるMSigDBです。MSigDBには、キュレーションされた遺伝子セット(KEGGのような)から、遺伝子オントロジー、制御モチーフ、文献によって定義された多くの単発のシグネチャーまで、多くの種類のパスウェイが含まれています。私たちは現在、MSigDBを用いた基本的なGSEA解析を微分発現解析パイプラインの一部として提供しており、将来的にはこれを拡張する予定です。
MSigDBは、研究対象の生物がヒトであれば素晴らしい選択肢ですが、ヒト以外の生物の場合はどうでしょう?MSigDBで定義されているパスウェイの大部分はヒトの文脈のものであり、酵母のような生物学は明らかに大きく異なります。しかし、MSigDBはマウスのようなヒト以外の生物種では、今でも頻繁に使われています。すでに述べたように、私たちはGSEA/MSigDBを発現変動解析パイプラインの一部として提供しており、生物種がヒトであろうとなかろうと提供しています。従って、ヒト以外の生物種を使用することがGSEA/MSigDBの統計的検出力にどのような影響を与えるかを調べたいと考えました。
結果
遺伝子の相同性情報のソースとしてNCBIのHomoloGeneを使用しました。HomoloGeneにはヒト、マウス、酵母など21種の情報が含まれています。図1は、ヒトとホモログを持つ遺伝子の数を生物種ごとに示したものです。チンパンジーのPan troglodytesやマウスのMus musculusなど、ヒトに近縁な種ほど、ヒトとの相同遺伝子の数が多いです。
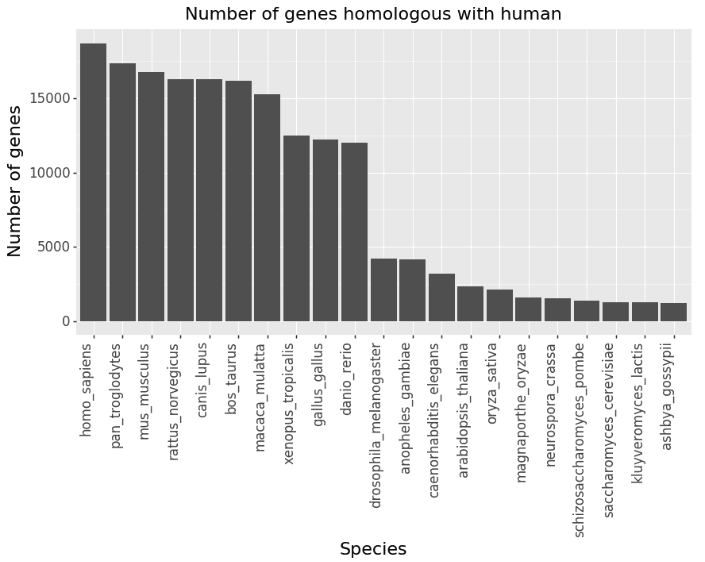
図1:ヒトとホモログを持つ各生物種の遺伝子数。左側にヒトのHomoloGeneにおける遺伝子数がプロットされています。
次に、MSigDBのパスウェイ(ヒトの遺伝子を使用)をヒト以外の生物種に変換した場合の影響を調べます。一つ注意すべきことは、GSEAには統計的な理由で小さすぎる(15遺伝子以下)、または大きすぎる(500遺伝子以上)パスウェイを削除するデフォルトオプションがあることです。図2は、ヒトの遺伝子シンボルを各生物種の遺伝子シンボルに変換した後、MSigDBに残ったパスウェイの数を示しています。図3は各生物種のパスウェイのサイズ分布の箱ひげ図です。
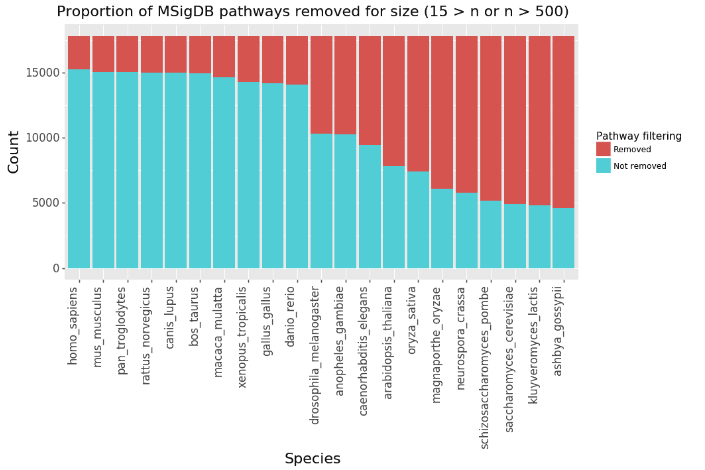
図2:各生物種について、MSigDBパスウェイ中の遺伝子がヒトとのホモログを持たない場合は削除されます。このプロットは、小さすぎる(15遺伝子未満)、または大きすぎる(500遺伝子以上)ために削除されたパスウェイの数を示しています。
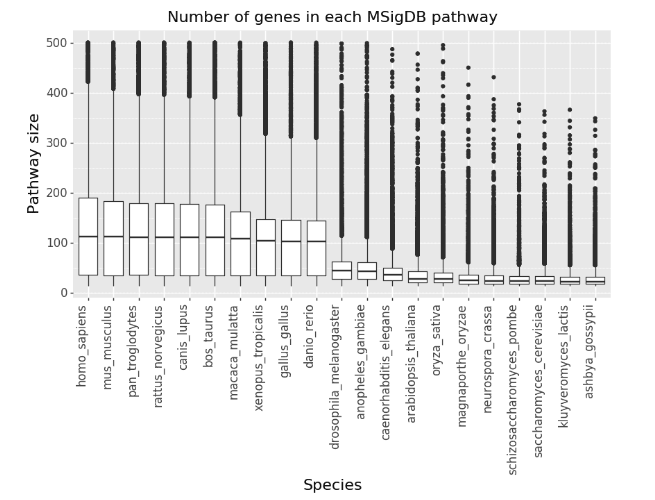
図3: それぞれの生物種について、MSigDBのパスウェイに含まれる遺伝子がヒトとのホモログを持たない場合は削除されます。プロットは、MSigDBの各パスウェイの遺伝子数のサイズ分布を示します。
次に、ヒト以外の生物種だった場合に、エンリッチされたパスウェイを検出するGSEAの統計的検出力への影響を評価しました。このような検証を行う理想的な方法は、各サンプルが同じ生物学的プロセスを摂動している21の生物種のRNA-SeqのデータがHomoloGeneにあり、これらを比較することです。この場合、各生物種のGSEA結果は相対的に同じになるはずです。しかし、そのような検証を行うのは、かなり困難か不可能です。例えば、MSigDBのパスウェイの多くで表現されているバイオロジーは、酵母のような遠縁の生物には存在しないかもしれません。しかし、私たちは、ヒトのRNA-seqデータでGSEAを行い、ある種の「真実のデータセット」とし、次にそれぞれの生物種について同じGSEAを行うことができます(同じヒトのデータセットを使います)。これにより、ヒトと他の生物種との間で相同性を欠く遺伝子を除去した場合に、「真実のデータセット」において同じパスウェイをどの程度同定できるかを調べることができます。
ヒト癌細胞株からのRNA-seqデータを用いてベンチマークを行いました[3]。6つのグループからそれぞれ無作為に10サンプルを選択しました:
- 乳房、腺がん
- 大腸・直腸、腺がん
- 肺、腺がん
- 脳、神経膠腫
- 胃、癌腫
- 卵巣、がん腫
次に、「乳房、腺がん」を他の5つのグループそれぞれと比較するGSEAを行いました。さらに、MSigDBの以下のパスウェイセットを用いて5つの比較を行いました:
- h.all.v6.2.entrez – ホールマーク遺伝子セット
- c2.all.v6.2.entrez – キュレーションされた遺伝子セット(KEGGなど)
まず、全遺伝子を用いてGSEAを行い(ヒトのサンプルで行うように)、パスウェイの「真実」セットを作成します。次に、その後の生物種ごとに、ヒトと相同性を持たない遺伝子をMSigDBから取り除いてから、再度GSEAを行います。精度は「真実」セットにも含まれるパスウェイの割合です。図4に解析結果を示します。驚くことではありませんが、GSEA解析の精度は遠縁の生物ほど下がるが、マウスやチンパンジーのような近縁の生物では問題ありません。しかし、「c2.all.v6.2.entrez 」パスウェイ・セットへの影響ははるかに小さいです。
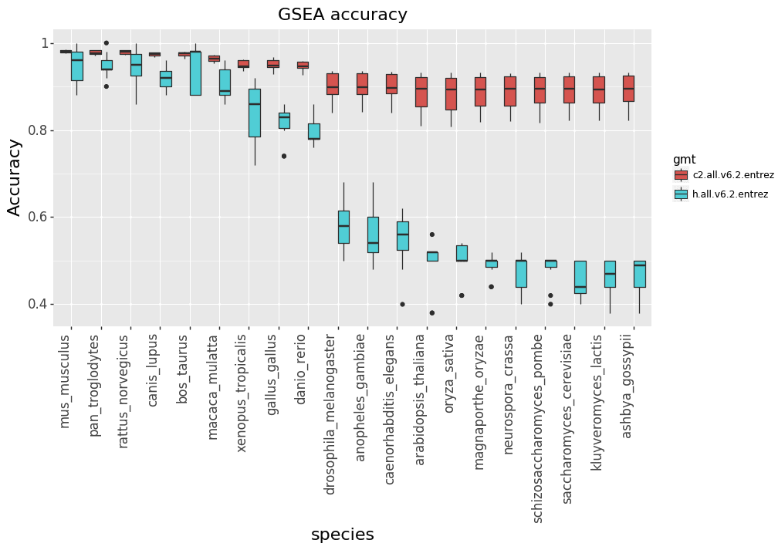
図4:各生物種について、MSigDBパスウェイ中の遺伝子がヒトとのホモログを持たない場合は削除されます。その後、フィルタリングされたMSigDBパスウェイを用いてGSEAが実行されます。精度は、遺伝子のフィルタリングを行わずにGSEAを実行した場合の結果と比較することで計算されます。
結論
Broad InstituteのGSEAとMSigDBパスウェイデータベースは、パスウェイ解析のための最も一般的なリソースの一つです。ほとんどのパスウェイ解析ソフトウェア(GSEA/MSigDBを含む)はヒトのサンプルを対象としています。ここでは、MSigDBのヒト遺伝子を非ヒト遺伝子に変換した場合の影響を評価しました。主な結果は以下の通りです:
- MSigDBパスウェイのヒト遺伝子をヒト以外の遺伝子に変換すると、遺伝子セットのサイズと数が大幅に減少します。しかし、マウスやチンパンジーのようなより近縁な生物種では、これはあまり当てはまりません。
- ヒト以外の生物種に変換した後にMSigDBでGSEAを実行すると、より遠縁の生物種で精度が著しく低下します。
参考文献
- Zyla J, Marczyk M, Weiner J, Polanska J. Ranking metrics in gene set enrichment analysis: do they matter? BMC Bioinformatics. BioMed Central; 2017 May 12;18(1):256.
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005 Oct 25;102(43):15545–50.
- Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015 Mar;33(3):306–12.
最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。