
このパイプラインを実行すると、最初に「ATAC-seq alignment (Bowtie2)」パイプラインが自動的に実行されます。その次に実行されるMACS2パイプラインでは、「ATAC-seq alignment (Bowtie2)」パイプラインで出力された重複リード除去済みのBAMファイルを使用します。ここでは、ピークコーリングとピークアノテーションが実行されます。そして最後に、HOMERを使用してエンリッチメント解析とモチーフ解析を行います。
パイプラインのワークフロー
BasepairでのMACS2パイプラインのワークフローを図1に示します。

ピークコーリング
MACS2でシーケンシングリードがエンリッチされたゲノム領域を同定します。これらの領域は、アクセス可能なクロマチン領域(オープンクロマチン領域)を示しています。
ピークアノテーション
MACS2で同定されたピークを、HOMERを用いて特定のゲノム領域に関連付けます。例えば、遺伝子、プロモーター、エンハンサーなどが含まれます(1)。
エンリッチメント解析
ピークアノテーションで関連付けられた遺伝子リストを、HOMERを用いて解析します。これは、オープンクロマチン領域に関連する遺伝子の役割を特徴付けるのに役立ちます。また、遺伝子が関与する生物学的プロセスについて、より深い情報を提供します(1)。
モチーフの探索とアノテーション
モチーフとは、アクセス可能なクロマチン領域内で繰り返し現れる短いDNA配列のことです。これらのモチーフは転写因子や他の制御タンパク質の結合部位に該当します。ここでは、統計的に過剰に出現しているモチーフを、HOMERを用いて同定します。さらに、エンリッチされたモチーフは、既知のモチーフにアノテーションされます(1)。
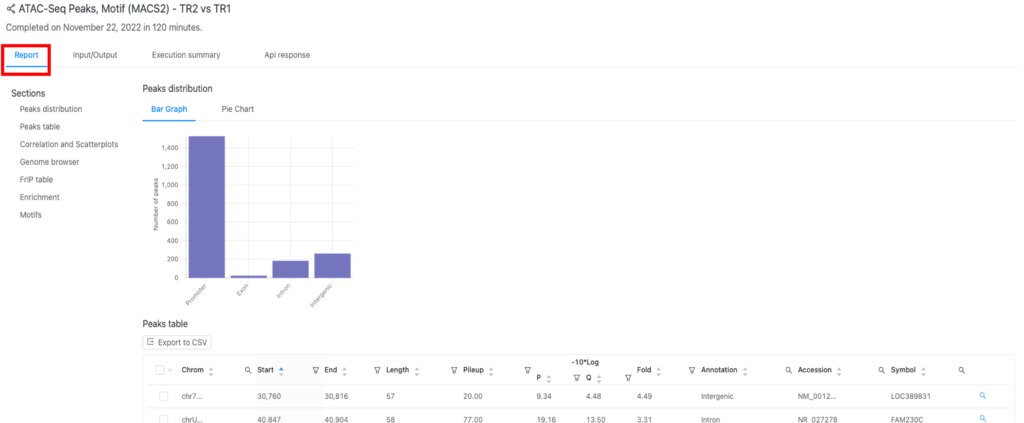
結果(「Report」タブ)
結果は「Report」タブ(図2の赤枠)にあります。
Peaks distribution
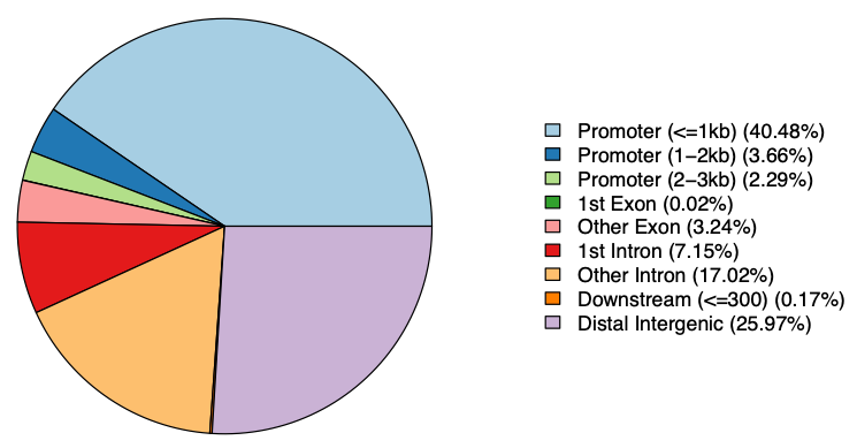
異なるゲノム領域で検出されたピークの分布を図3に示します。
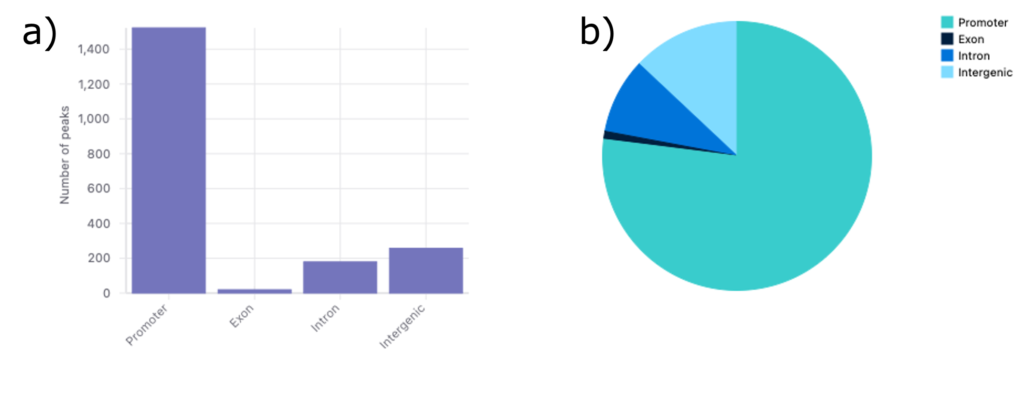
棒グラフ(図3a)では、最も多くのピークがプロモーター領域に集中していることがわかります。これは、プロモーター領域が非常にアクセスしやすいことを示しており、プロモーターが転写開始において重要な役割を担うことから予想される結果です。
また、遺伝子間領域やイントロン領域においては多くのピークが見られ、これらの領域にもアクセス可能なクロマチンが存在することを示唆しています。これは、サンプル中に制御エレメントが存在する可能性を示しています。一方、エクソン領域におけるピークは最も少なく、コード領域へのアクセシビリティが低いことを示しています。
円グラフの結果(図3b)は棒グラフの結果と一致しています。
Peaks table
同定されたピークとその近傍遺伝子のアノテーションを図4に示します。
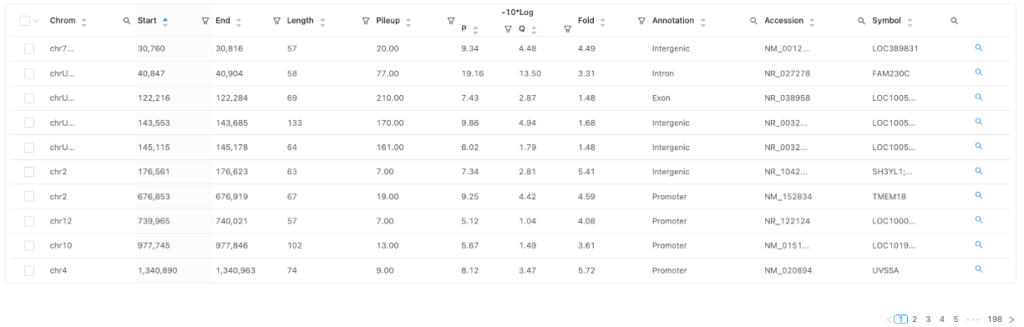
用語
| 用語 | 定義 |
| Chrom, Start, End, Length | 染色体番号、ゲノム上の位置とピークの大きさ |
| Pileup | ピーク頂上のゲノム位置にアライメントされたリード数 |
| P | ピークの統計的な有意性( -10 * log10 ) |
| Q | 偽発見率(FDR)で補正した統計的な有意性( -10 * log10 ) |
| Fold | ピークのエンリッチメント度。バックグラウンドに対してどれくらいエンリッチしているかを表す |
| Annotation | ピークが位置するゲノムの特徴 |
| Accession | ピークに最も近い遺伝子のID |
| Symbol | ピークに最も近い遺伝子の記号・名前 |
Correlation and scatterplots
相関解析から得られたヒートマップと散布図をそれぞれ図5aと5bに示します。
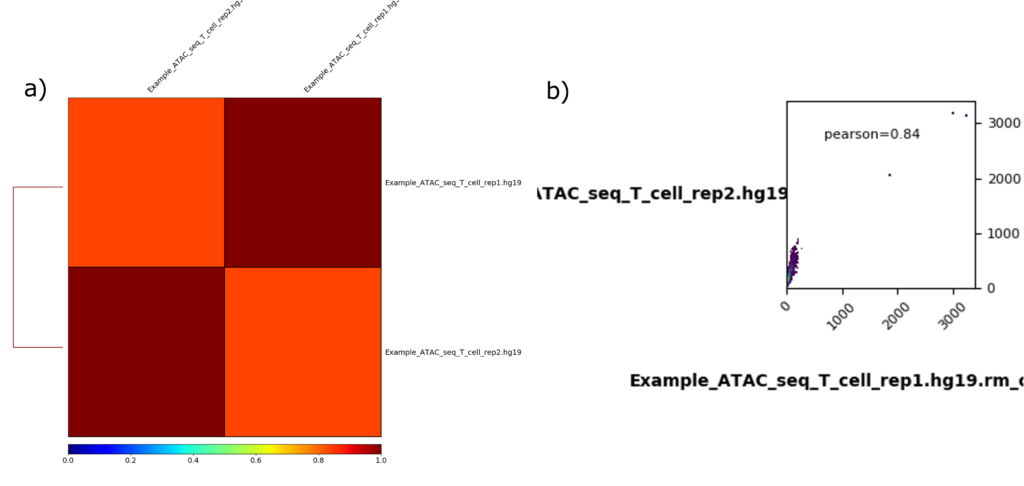
ヒートマップ
ヒートマップ(図5a)では、x軸とy軸の異なるサンプルの相関が比較されています。また、ヒートマップの下部にあるカラースケールは、相関のレベルを示しています。赤色(値が1に近い)は強い相関を示します。つまり、2つのサンプルのシグナルプロファイルが非常に類似していることを示します。一方、青色(値が0に近い)は相関が弱いことを示します。つまり、2つのサンプルの間にはほとんど類似性がないことを示します。
サンプル間のヒートマップの色の違いは、両者に中程度の相関があることを示しています。すなわち、図5aでは2つのサンプルデータが非常に類似していることを示していると同時に、再現性の良さを反映しています。
散布図
散布図(図5b)では、x軸はT cell rep 1のシグナル強度を表します。また、y軸はT cell rep 2のシグナル強度を表します。各ポイントは、特定のゲノム上の位置(例えば、ピークやターゲット領域)における2つのサンプル間の比較を表します。各点の位置は、一方のサンプルのシグナル強度(例えば、リードカウントまたは正規化スコア)が他方のサンプルのシグナル強度とどのように関連しているかを示します。
図5bのプロットには、2つのサンプル間の相関を測定するPearson correlation coefficient数値(pearson = 0.84)が表示されています。通常、係数は-1から1の範囲で、1は完全な相関、0は相関なし、-1は完全な負の相関を表します。左下の隅(原点近辺)に密集したプロットは、両方のサンプルでシグナル強度が低いゲノム領域が多いことを示しています。Pearson correlationの0.84は、2つのサンプル間に正の相関があることを示しており、シグナルが一致していることがわかります。
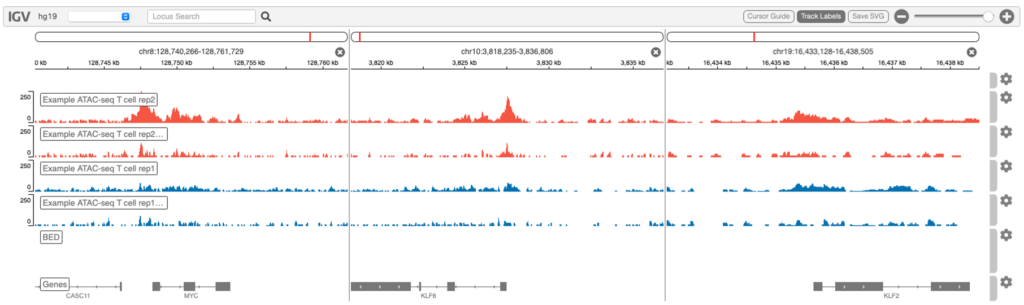
Genome browser
Integrative Genomics Viewer(IGV)でシグナル強度を視覚化した例を図6に示します。
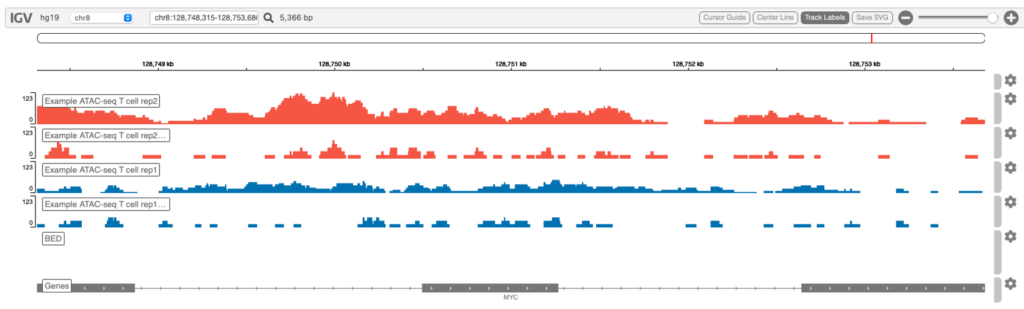
検索ボックスで、間にスペースを入れて複数の遺伝子を指定することで、並べて可視化することができます。例えば:

FRiP table
検出されたピークは、ゲノム中でリードが有意にエンリッチされた領域であり、オープンクロマチン領域を示しています。FRiP(Fraction of Reads in Peaks)スコアは、実験の質を評価するために計算され、オープンクロマチン領域として同定されたピーク内にマップされたシーケンシングリードの割合です。ピークにマップされたリード数をリードの総数で割ることで算出されます:
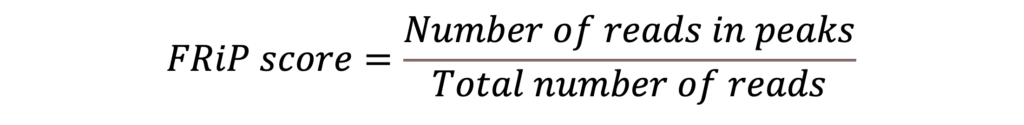
FRiPスコアが高いほど、オープンクロマチン領域のピークにマップされたリードの割合が高くなります。つまり、データから得られたクロマチンのアクセシビリティに関する情報の信頼性が高まります。解析に使用した2つのサンプルのFRiPスコアを図7に示します。

図7では、T cell rep1とT cell rep2のFRiPスコアはそれぞれ0.0007と0.003でした。つまり、サンプルT cell rep1およびT cell rep2の全リードのうち、それぞれ0.07%および0.3%のみがピークに含まれることを示しています。
高品質のATAC-seqデータのFRiPスコアは通常0.2(20%)以上ですが、これはサンプルや実験条件によって異なる可能性があります。
Enrichment
ピークに関連する遺伝子のエンリッチメント解析の結果を図8に示します。
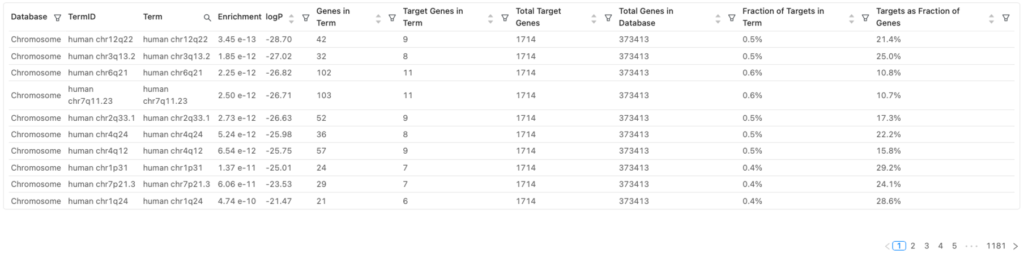
用語
| 用語 | 定義 |
| Database | エンリッチ解析で使用される遺伝子セットのデータベース |
| TermID | 遺伝子セットのデータベースにおけるユニークな識別子 |
| Term | 遺伝子セットの名前 |
| Enrichment logP | 遺伝子セットのエンリッチメントの有意性(対数変換したp値) |
| Genes in Term | 遺伝子セットに含まれる遺伝子の総数 |
| Target Genes in Term | データセットと遺伝子セットの間で共通の遺伝子の数 |
| Total Target Genes | データセットに含まれる遺伝子の総数 |
| Total Genes in Database | 遺伝子セットのデータベースに含まれる総遺伝子数 |
| Fraction of Targets in Term | データセットに含まれる全遺伝子の内、遺伝子セットで共通する遺伝子の割合(Target Genes in Term / Total Target Genes) |
| Targets as Fraction of Genes | 遺伝子セットに含まれる全遺伝子の内、データセットに含まれる遺伝子の割合(Target Genes in Term / Genes in Term) |
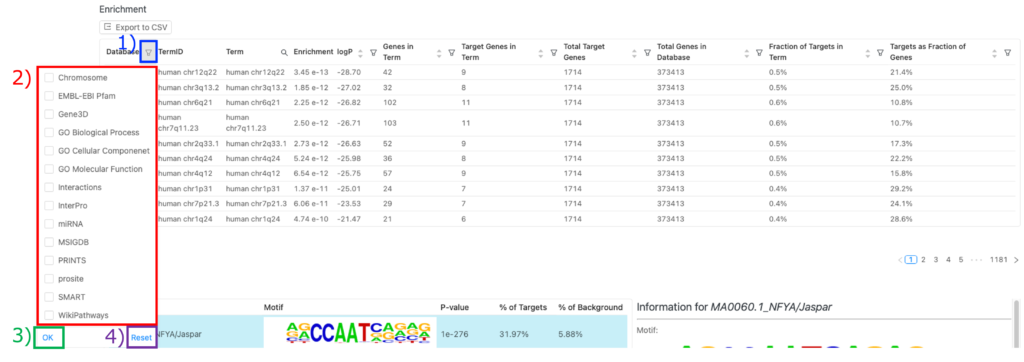
エンリッチメント解析結果を表示するデータベースの変更
1) Enrichment 表の「Database」タブにある「filter」アイコンをクリックしてください。
2)データベースをチェックしてください。
3) 「OK」をクリックしてください。
4) デフォルトのデータベース(例:Chromosome)に戻りたい場合は、「Reset」をクリックし、チェックしたすべてのデータベースのチェックを外してください。
Motifs
ピークで同定され、エンリッチされたモチーフの要約と、それらにマッチする転写因子結合プロファイル を図9に示します。

用語
| 用語 | 定義 |
| Best Match | 発見されたモチーフに最も類似した既知のモチーフまたは転写因子結合プロファイル |
| Motif | 発見されたモチーフの配列ロゴ。背の高い文字は、その位置のヌクレオチドの保存性が高いことを示します |
| p-value | エンリッチメントの統計的有意性 |
| % of Targets | モチーフを含むピーク領域(ターゲット)の割合 |
| % of Background | モチーフを含むバックグラウンド領域(非ピーク領域)の割合 |
図9の左側のモチーフ(例えば、NFYA転写因子のベストマッチ)をクリックすると、そのモチーフの詳細なサマリーが右側に表示されます。図9のエンリッチされたモチーフの詳細については、テクニカルノート「Basepairでのエピジェネティクスデータのモチーフ探索とアノテーション」をご参照ください。
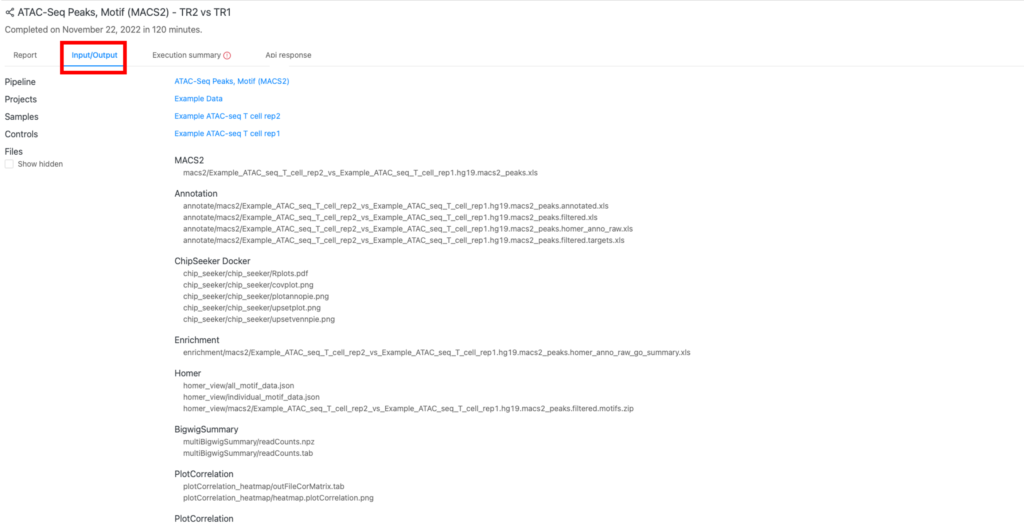
アウトプットファイル (「Input/output)タブ)
アウトプットファイルは、「Input/output」タブ(赤枠)にあります。
MACS2
- ピークコールの生データ(Excel形式)
(macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.xls)
Annotation
- 近傍遺伝子にアノテーションされたピークのリスト(Excel形式)
(annotate/macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.annotated.xls)
- ブラックリストに載った領域を除いたピークのリスト(Excel形式)
(annotate/macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.filtered.xls)
- HOMERで生成されたピークとそのアノテーションのリスト(Excel形式)
(annotate/macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.homer_anno_raw.xls)
- HOMERでフィルタリングされたピークとそのアノテーションのリスト(Excel形式)
(annotate/macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.filtered.targets.xls)
ChipSeeker Docker
- 特定の遺伝子要素に近接したピークの割合を示す円グラフ(PDF形式)
(chip_seeker/chip_seeker/Rplots.pdf)
- 各染色体上で見つかったピークを可視化したグラフ(PNG形式)
(chip_seeker/chip_seeker/covplot.png)
- 特定の遺伝子要素に近接したピークの割合を示す円グラフ(PNG形式)
(chip_seeker/chip_seeker/plotannopie.png)
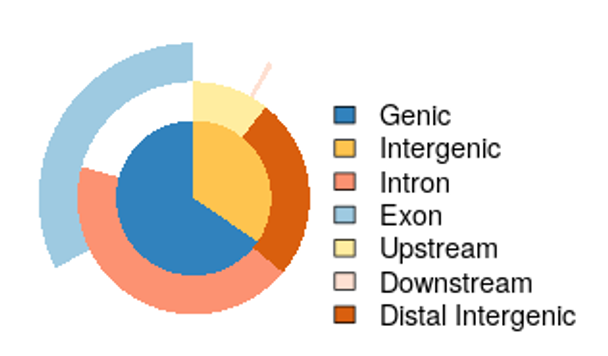
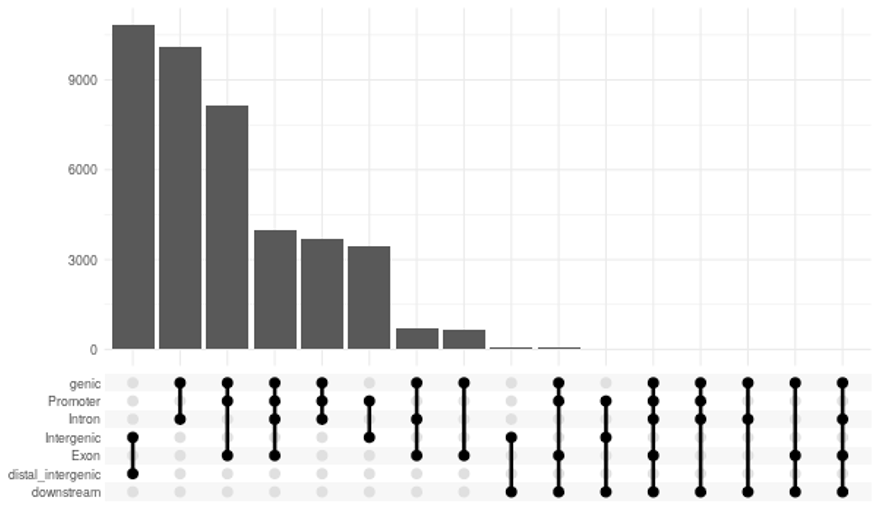
- 特定の遺伝子要素に近接したピークのアップセットプロット(PNG形式)
(chip_seeker/chip_seeker/upsetplot.png)
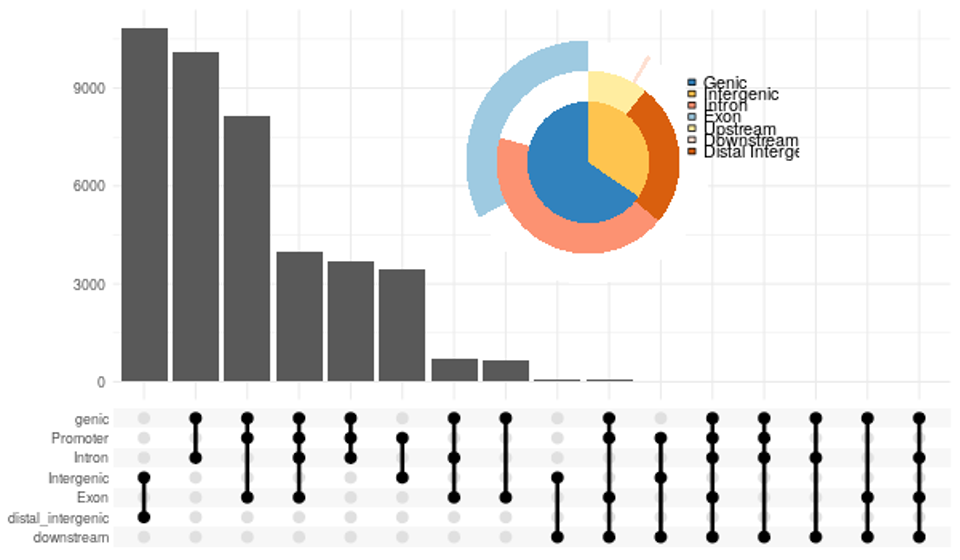
- 円グラフとアップセットプロットを組み合わせたプロット(PNG形式)
(chip_seeker/chip_seeker/upsetvennpie.png)
Enrichment
- HOMERによる遺伝子オントロジーエンリッチメント解析の結果(Excel形式)
(enrichment/macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.homer_anno_raw_go_summary.xls)
Homer
- 解析で同定されたすべてのモチーフのデータ(JJSON形式)
(homer_view/all_motif_data.json)
- 解析で同定された個々のモチーフのデータ(JJSON形式)
(pagehomer_view/individual_motif_data.json)
- HOMERの解析結果の圧縮フォルダ
(pagehomer_view/macs2/<ANALYSIS_NAME>.<genome>.macs2_peaks.filtered.motifs.zip)
BigwigSummary
- 解析に使用した全サンプルのピークの正規化されたリードカウントデータ(NPZ形式)
(multiBigwigSummary/readCounts.npz)
- 解析に使用した全サンプルのピークの正規化されたリードカウントデータ(タブ区切り形式)
(formatmultiBigwigSummary/readCounts.tab)
Plot Correlation
- 異なるサンプル間の相関を示したマトリックス(タブ区切り形式)
(plotCorrelation_heatmap/outFileCorMatrix.tab)
- 異なるサンプル間の相関を示したヒートマップ(PNG形式)
(plotCorrelation_heatmap/heatmap.plotCorrelation.png)
- 異なるサンプル間の相関示した散布図(PNG形式)
(plot Correlation_scatterplot/scatterplot.plotCorrelation.png)
参考文献
1. Peak calling with MACS2 | Introduction to ChIP-Seq using high-performance computing
関連ブログ
- ATAC-seq解析 https://basepairtech.jp/analysis/atac-seq/
- Basepairでのエピジェネティクスデータのモチーフ探索とアノテーション https://basepairtech.jp/blog/2230/