
deFuseパイプラインは遺伝子融合を検出するために使用されます。このパイプラインはまずリードをリファレンスゲノムにアライメントします。次に、融合している可能性のある遺伝子を検索します。最後にフィルタリングを行い、偽陽性の融合遺伝子を除去します。
遺伝子融合の発見が重要な理由
遺伝子融合とは、2つの別々の遺伝子が異常に結合する現象です。これはゲノムの構造的な再配列に起因する可能性があり、しばしば癌で観察されます。遺伝子融合は異常な融合タンパク質の生成につながります。これらは遺伝子発現の調節異常を引き起こす可能性があります。したがって、これらの融合遺伝子を同定することは、特に癌研究において極めて重要です。
パイプラインのワークフロー
図1に、deFuseパイプラインのワークフローを示します。

アライメント
deFuseはRNA-seqリードを取得し、リファレンスゲノムにアライメントします。
スプリットリード検出
アライメント後、deFuseは「スプリットリード」を探します。これは部分的に異なる位置にアライメントするリードです。これらのリードはしばしば融合境界にまたがっており、遺伝子融合の直接的な証拠となります。例えば、1つのリードが部分的に遺伝子Aまたは遺伝子Bにアライメントすることがあります。
遺伝子融合の同定
遺伝子融合の可能性が特定されると、deFuseは融合遺伝子候補のリストを作成します。スコアリングシステムを用いて、最も可能性の高い遺伝子融合に優先順位をつけます。
フィルタリング
deFuseは、偽陽性を除去するために一連のフィルターを適用します。例えば、反復配列のような相同性の高い領域から生じる融合や、ミスアライメントから生じる融合などを除去します。
結果(「Report」タブ)
すべての結果は「Report」タブ(図2の赤枠)にあります。
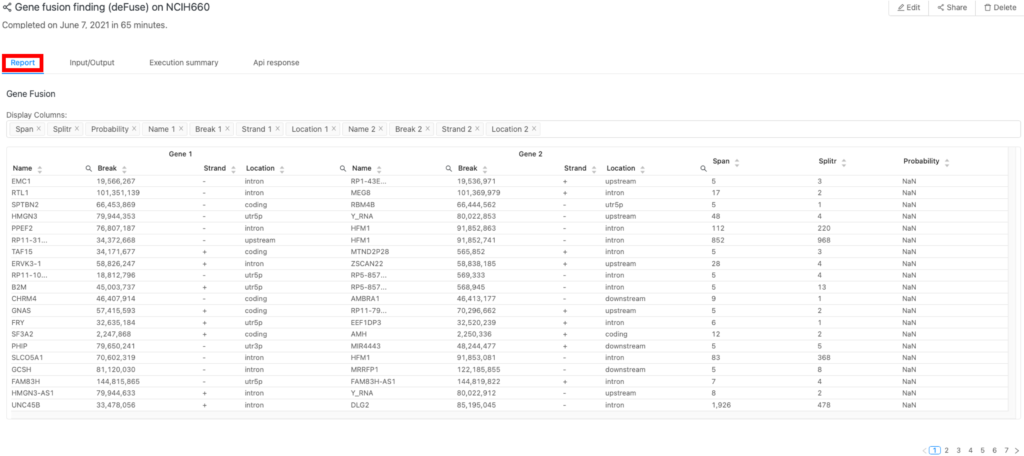
Gene fusion
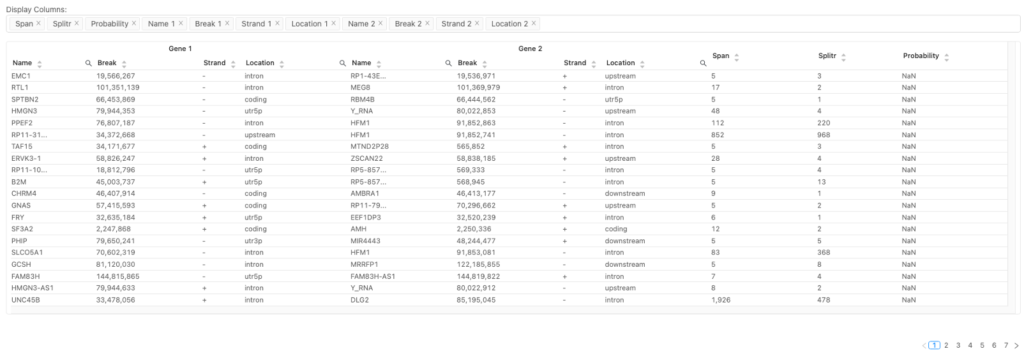
図3は、同定された遺伝子融合とその詳細を示しています。例えば、スプリットリードとスパニングリードの数が多いほど、本物の融合である可能性が高いことを示しています。遺伝子の位置に関する詳細は、遺伝子機能に対する各融合の潜在的な影響を評価するのに役立ちます。これらの情報は癌研究、特に融合が癌化イベントの原因であるかどうかを判断するのに重要です。
用語
| 用語 | 定義 |
| Gene 1 & 2 | 融合に関与する2つの遺伝子 |
| Name | 遺伝子名 |
| Break | 融合に関与する各遺伝子の染色体上のブレイクポイント位置 |
| Strand | 融合に関与する遺伝子の方向 |
| Location | 各遺伝子上の融合位置。例えば: intron:融合はイントロン(非コード領域)内で発生。 coding:融合はタンパク質コード領域を阻害。 utr5p:融合は5’または3’非翻訳領域(UTR、untranslated regions)に影響。これらの領域は遺伝子発現制御に関与しますが、タンパク質のコードには関与しません。 Locationは、遺伝子機能に対する各融合の潜在的な影響を評価するのに有用です |
| Span | スパニングリードの数。つまり、融合境界で分割せずにアライメントしたリード |
| Splitr | スプリットリードの数。すなわち、融合ブレイクポイントで2つの遺伝子の間で分割されたリード |
| Probability | 各融合イベントの信頼度スコア。「NaN」は未計算を表します |
アウトプットファイル(「Input/output 」タブ)
アウトプットは、「Input/output 」タブ(図4の赤枠)にあります。

deFuse
| ファイル | 説明 |
| defuse/<sample>.results.tsv | 検出された融合イベントのリスト(TSVファイル) |
| defuse/<sample>.results.classify.tsv | 融合イベントと分類情報を含むTSVファイル。分類は、信頼度の高い融合と偽陽性を区別するのに役立ちます。分類は確率(信頼度スコア)を割り当てることによって行われます |
| defuse/<sample>.results.filtered.tsv | 信頼度スコアでフィルタリングされた融合イベントを含むTSVファイル |
参考文献
関連ブログ
BasepairのRNA-Seq https://basepairtech.jp/analysis/rna-seq/