
なぜFASTQの品質が重要か
FASTQの品質は、下流の解析の品質に影響します。
FASTQデータには、低品質リードが含まれることがあります。多くの場合、これはシーケンシングエラーやPCRバイアスなどの要因によって生じます。また、シーケンスプロセス中、アダプター配列がリードの末端に付加されます。アダプター配列はシーケンスには必須です。しかし、ゲノム内の領域にはマッピングされません。
これらは、ダウンストリーム解析の妨げとなり、不正確な結果につながります。そのため、品質評価(QC)、トリミング、フィルタリングは、重要なステップです。これらの処理を適切に行うことにより、正確で信頼性の高い結果を生成できます。結果として、アライメントの効率も向上します。
FASTQの品質評価(QC)および処理ワークフロー
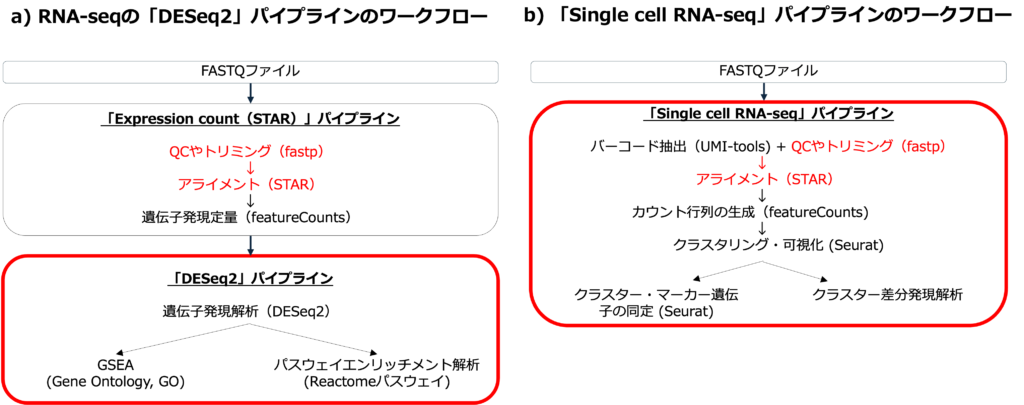
RNA-seqのDESeq2(図1a)とシングルセルRNA-seqパイプライン(図1b)のワークフロー例を示します。どちらのパイプラインでも、Expression Count (STAR)パイプラインは事前に自動的に行われます。
Basepairでは、QCやアライメントを別途実行する必要はありません。
FASTQの品質評価とアダプタートリミング
トリミングはfastpで、リードから低品質のリードとアダプターを除去します。まず、低品質のリードは、最小品質閾値を設定することで除去されます。次に、この閾値以下の塩基が除去されます。ライブラリー調製時に付加されたアダプター配列は、自動的に検出され除去されます。
リードのフィルタリング
トリミング後に短すぎるリードは除去されます。このステップにより、高品質のリードのみがダウンストリーム解析に使用されます。
アライメント
リファレンスゲノムに対するリードのアライメントは、STARで実行されます。ここでは、リードがどこから来たかを決定します。アライメントプロセスでは、まずリファレンスゲノムのインデックスを作成します。その後、リードをリファレンスゲノムにアライメントします。
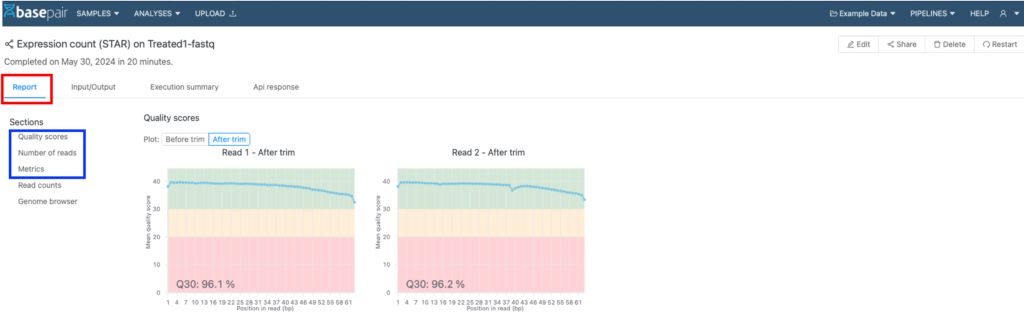
結果の表示:「Report」タブ
FASTQの品質結果は「Report」タブ(図2の赤枠)に表示されます。「Report」タブには、以下ののセクション(図2の青枠)があります。
- Quality score
- Number of reads
- Metrics
Quality Score
Q Scoreの計算
品質評価(QC)では、生のリードの品質を評価します。Basepairでは、fastpを使用してこれを行います。QCでは、読み取りの各塩基にQ scoreを割り当てられます。Q scoreは、その塩基が誤ってコールされる確率の負の対数を表します。
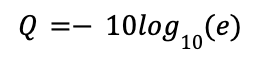
Q scoreが高いほど、誤りの確率は小さくなります。
クオリティプロットの見方
通常、Q scoreはQ10、Q20、Q30に分類されます。配列決定品質スコアとコールされた塩基の精度の関係を表1に示します。例えば、Q10の場合、エラー率は10分の1です。これは、10bpのリードのうち1本にエラーが含まれる可能性があることを表します。一方で、Q30の場合、ほぼすべてのリードにエラーがなく、正しい状態です。
| Q score | Probability of incorrect base called | Accuracy of base called | |
| 10 (Q10) | 品質良好(緑) | 1 in 10 | 90% |
| 20 (Q20) | 妥当な品質(黄) | 1 in 100 | 99% |
| 30 (Q30) | 品質不良(赤) | 1 in 1000 | 99.9% |
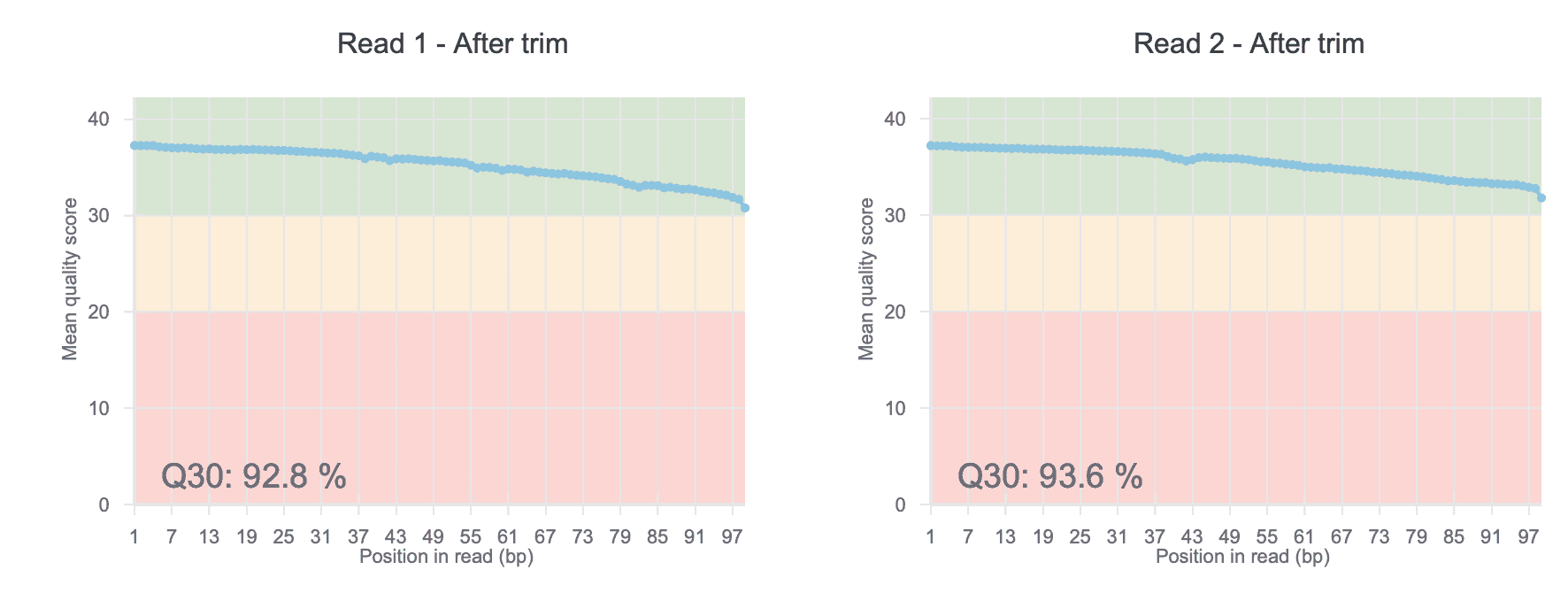
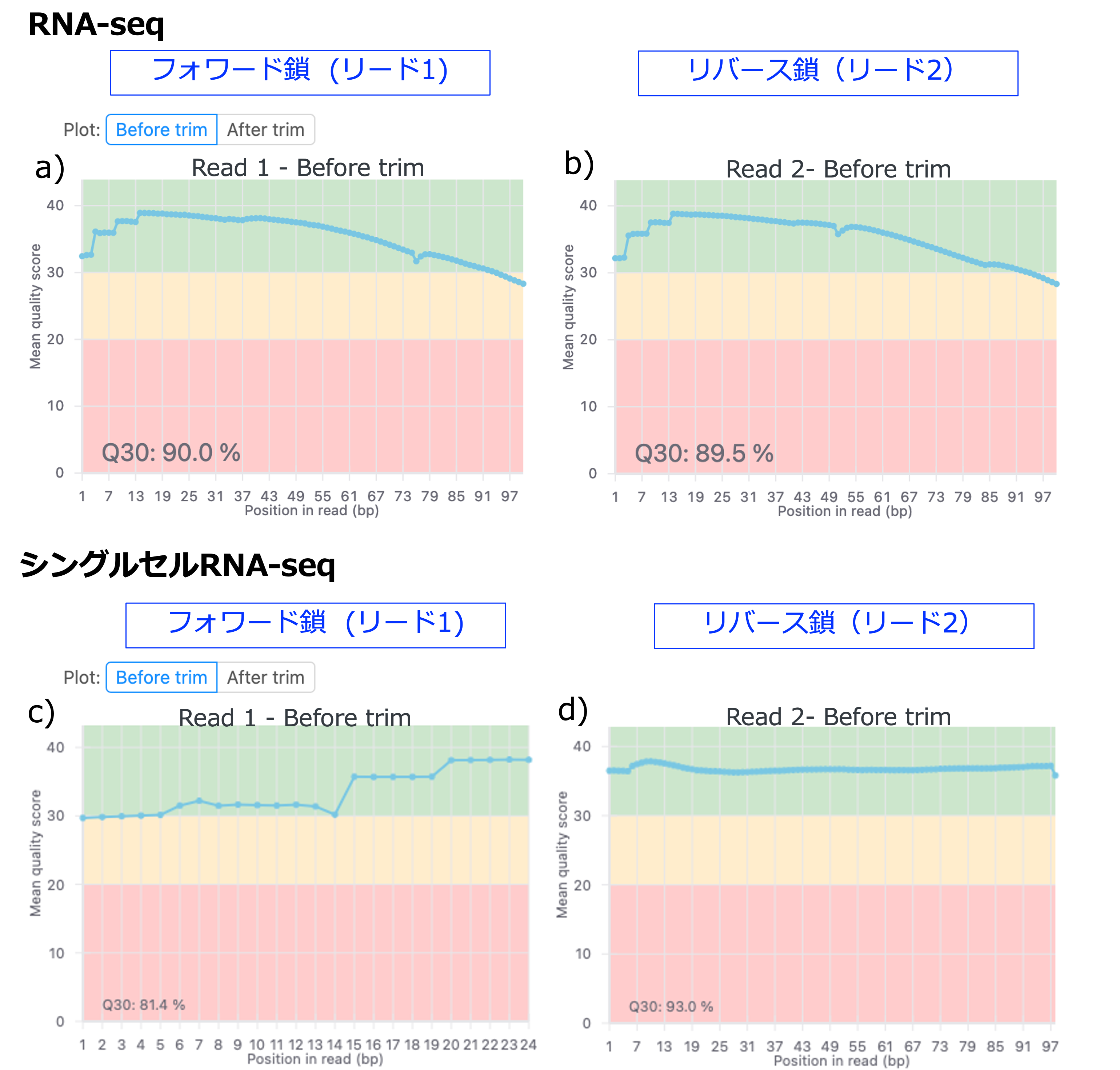
クオリティとアダプタートリミング前後の塩基あたりの平均Q scoreは、塩基あたりの配列クオリティプロットで示されます。例えば、RNA-seqデータでは、Q score30のリードがフォワード鎖(リード1)に90.0%あり(図3a)、トリミング前のQ score30のリードがリバース鎖(リード2)に89.5%あります(図3b)。
Number of reads
マップされたリードのパーセンテージをSankeyプロットで示します。Sankeyプロットの例を図4に示します。マップされたリードの割合は、全体的なシーケンス精度とコンタミネーションDNAの有無を示す重要な指標です。通常のRNA-seqおよびscRNA-seqリードの70%から90%はリファレンスゲノムとアライメントし、ほとんどのリードはユニークアライメントされると予想されます。
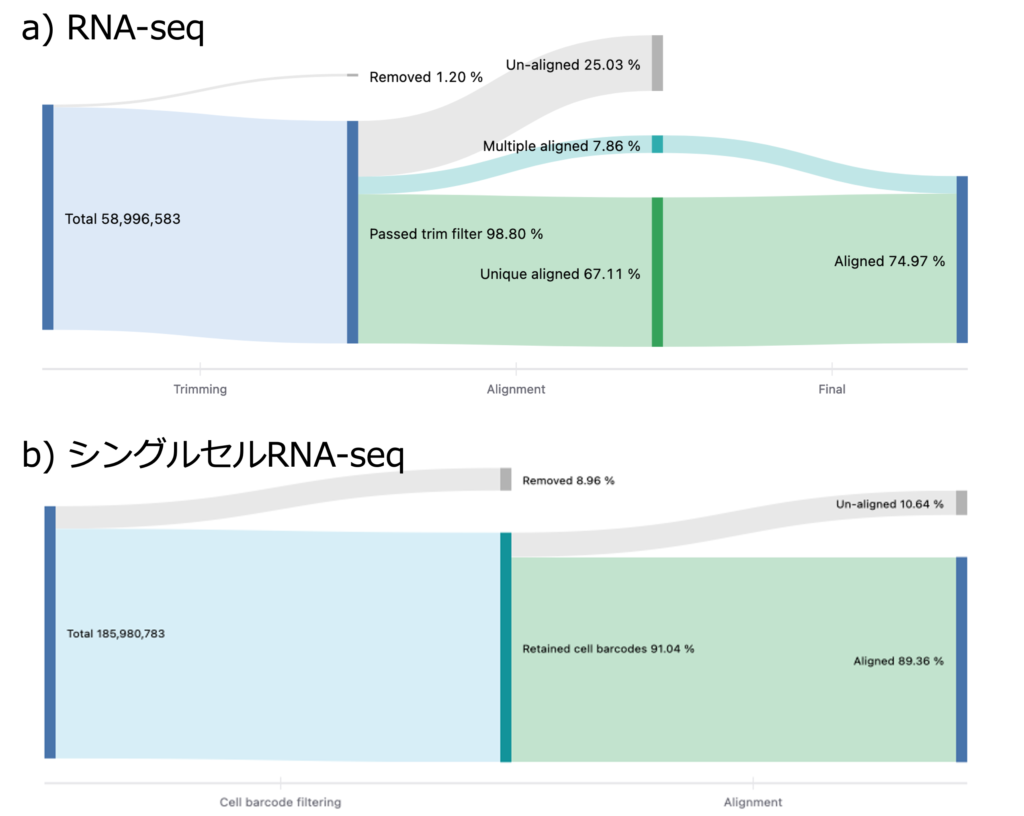
図4. QCとアライメント前後のSankeyプロット。
Sankey plotで使用される用語
| 用語 | 定義 |
| Total | トリミング前のアライメントに使用された入力リード数の合計。「Passed trim filter」と「Removed」の合計 |
| Passed trim filter | 「Total」からのリードのうち、トリムフィルターを通過したリードの割合。「Unique aligned」、「Multiple aligned」、および「Un-aligned」の合計 |
| Removed | 「Total」からのリードのうち、トリム フィルターを通過せず削除されたリードの割合 |
| Unique aligned | 「Passed trim filter」からのリードのうち、リファレンスゲノム内の 1 つの場所にのみマップされたリードの割合 |
| Aligned | リファレンスゲノムにアライメントされたリードの割合。「Unique aligned」と「Multiple aligned」の合計 |
| Un-aligned | リファレンスゲノムにアライメントされていないリードの割合 |
| Multiple aligned | 「Passed trim filter」からのリードのうち、リファレンスゲノムに複数回マップされたリードの割合 |
| Retained cell barcodes | 「Total」からの、バーコードが検出されたセルからのリードの割合 |
Metrics
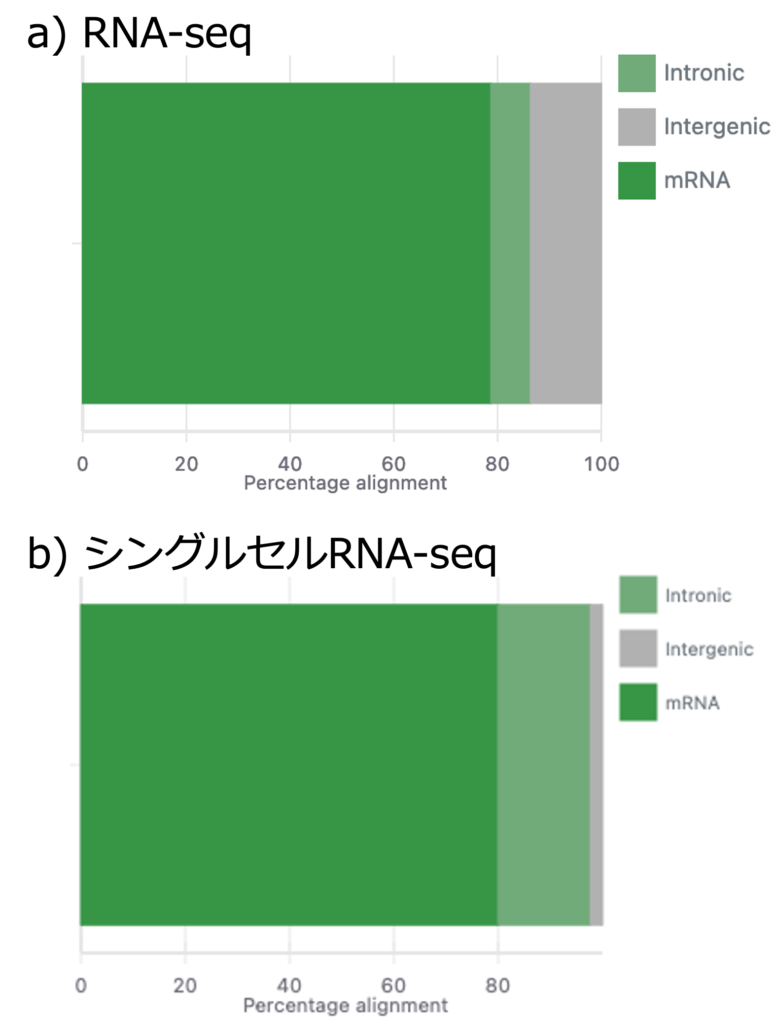
アライメントのサマリープロットを図5に示します。mRNA領域、イントロン領域、遺伝子間領域にアライメントされた割合を示しています。例えば図5aでは、79.34%のリードがエクソン領域(コーディングmRNA)、11.65%が遺伝子間領域、9.00%がイントロン領域にアライメントされています。
まとめ
品質評価(QC)、トリミングとフィルタリングは、データ解析前にFASTQファイル中の低品質リードやアダプター配列を除去するための重要な処理ステップです。Basepairのパイプラインでは、品質評価、トリミング、フィルタリングとアライメントはすべて最初に自動的に行われます。つまり、Basepairでこれらのステップが自動化されることで、下流の解析が容易になります。
参考文献
1. Illumina (2024). Measuring sequencing accuracy. Illumina. https://www.illumina.com/science/technology/next-generation-sequencing/plan-experiments/quality-scores.html
関連ブログ
RNA-seq解析 https://basepairtech.jp/analysis/rna-seq/
シングルRNA-seq解析 https://basepairtech.jp/analysis/single-cell-rna-seq/
最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。