

RNA-Seqのデータ解析は、エクセルやRと格闘したり、受注会社に依頼したりすることが多いと思います。これには、時間とコストがかかります。インタラクティブレポートを使うと、大きく時間をコストを圧縮することができます。
RNA-Seqとは?
RNA-Seqはサンプル、あるいはサンプル群の間で、変動している遺伝子を探すための手法です。パワフルであり、NGS解析の中で最も利用されているアプリケーションの一つです。多くの場合、サンプル間、あるいはサンプル群間で発現が変動している遺伝子(群)を見つけることを目的としています。
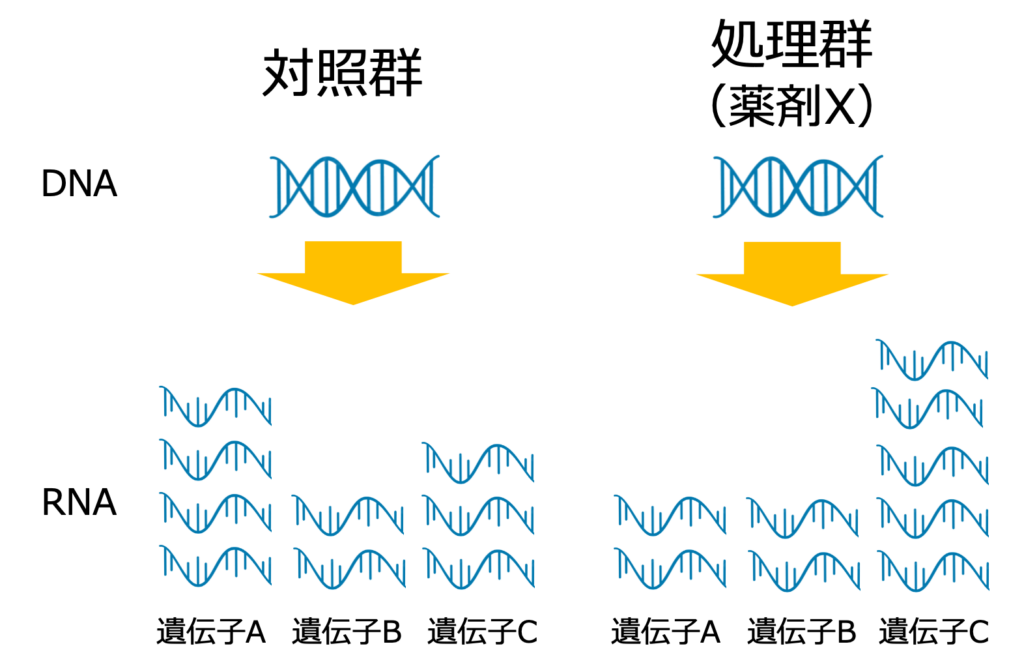
例えば、対照群=コントロール群と、処理群=薬剤Xで刺激をした群があるとします。右の例では、対照群では、遺伝子Aは4、遺伝子Bは2、遺伝子Cは3だったのが、処理群では、遺伝子Aは2、遺伝子Bは2、遺伝子5になったとします。つまり、遺伝子Aが減って、遺伝子Cが増えたということがわかります。
遺伝子は、一つで動いているわけではないので、周辺の遺伝子とグループ化されている場合があり、機能のグループをジーンオントロジー=GO反応経路をPathwayと言います。このケースの結果を言い換えると「薬剤Xは、遺伝子AとCもしくは、遺伝子AとCが関わっているGOとPathwayに影響を与えているということができます。
絞り込みができたら、より深く研究を進めていく、という流れになります。
RNA-Seqデータ解析の流れ
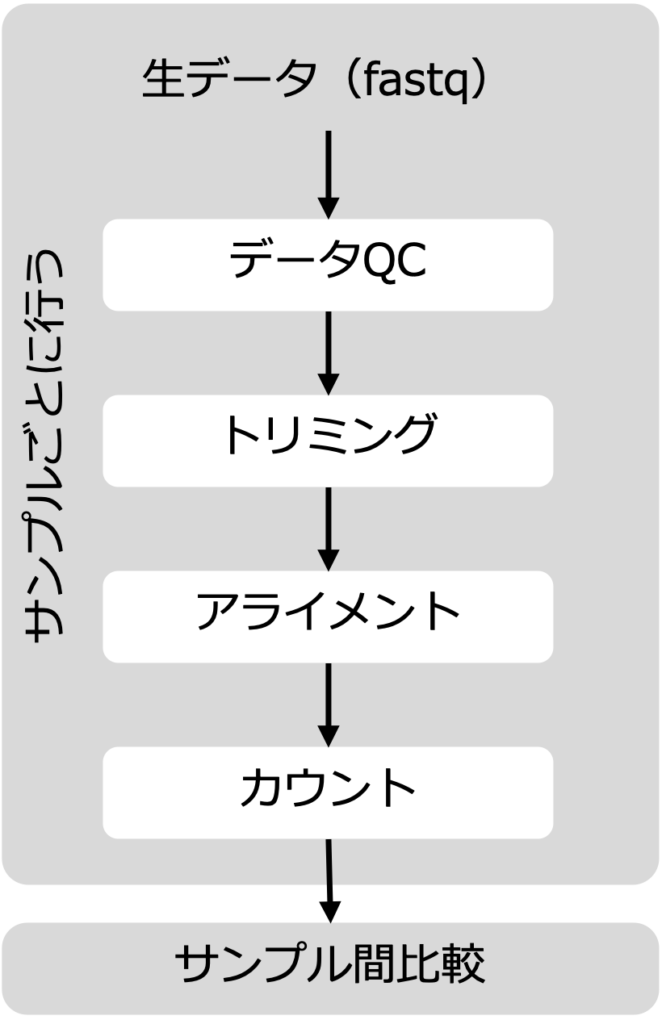
では、RNA-Seqのデータ解析の流れはどうなっているのでしょうか。
まずインプットは、fastqというフォーマットのNGSからの生データになります。このデータは、NGSから吐き出された配列情報がそのままの状態で記述されています。多くのRNA-Seqの場合、6Gb程度のサイズになります。テキストデータなので人間が読むことはできます。とはいえ、この状態では、変動している遺伝子を見つけるという目的が達成されません。そのため、人間が理解できるようにするプロセスが必要になります。
QCとトリミング
NGSシーケンスは、データにクオリティの低い部分がある場合があります。そのため、まずデータQCを行います。その後、アダプター配列やインデックス配列などを除くためのトリミングを行います。これでようやく、この後の解析に使えるデータになります。
アライメント(マッピング)とリードカウント
この状態では、まだACGTが並んだ配列情報、つまりリードでしかありません。どのリードがどの遺伝子に相当するかという情報は持っていません。この配列はこの遺伝子ですよ、とリードをゲノムに対して照合するステップをアライメント、もしくはマッピングと言います。
ようやく、どのリードがどの遺伝子かがわかったので、あとはカウントするだけです。
ここまでのプロセスはサンプルごとに行います。このアライメントが最も計算リソースを必要とするステップです。よく使われる「STAR」というパイプラインは、マルチコアのCPUが必要です。サンプル数が多い場合は、PCのリソースを数時間から数日占拠する程度の負荷がかかります。RNA-Seqの標準的なリード数が40M(4,000万)リードと考えると納得です。
比較
サンプルごとにリードカウントを行ったあと比較してやると、ようやく発現が変動している遺伝子を抽出することができます。「DESeq2」などのパイプラインが利用されます。DESeq2はRパッケージですので、手元のPCで行うためにはRが必要になります。このDESeq2を使うと、発現変動遺伝子のリストやRNA-Seqでよく使われるボルケーノプロットなどを得ることができます。
シーケンスのレポートを受け取ったあとにやること
多くの場合、NGSは受託企業に依頼している、あるいは共同研究相手に依頼するなどしていると思います。典型的なNGSの受託会社さんだとPDFのレポートとエクセルファイル、生データのfastqを受け取ることになります。PDFレポートには、QCの結果や発現変動解析のグラフ、プロットが載っています。発現カウントや変動遺伝子のリストはエクセルファイルになっています。一部、BAMファイルといってアライメント結果もくれるところもあります。
多くは、発現変動遺伝子のエクセルを使って、発現変動遺伝子や自分の見たい遺伝子を探していくことになります。
有意に差のある遺伝子の絞り込み
発現変動遺伝子のエクセルは、縦が遺伝子、横に情報が並んでいます。縦は遺伝子リストになっていますので数万行、横は数十行になる、大きなエクセルです。パソコンのスペックがそこそこないと、開くのすら苦労ししそうです。
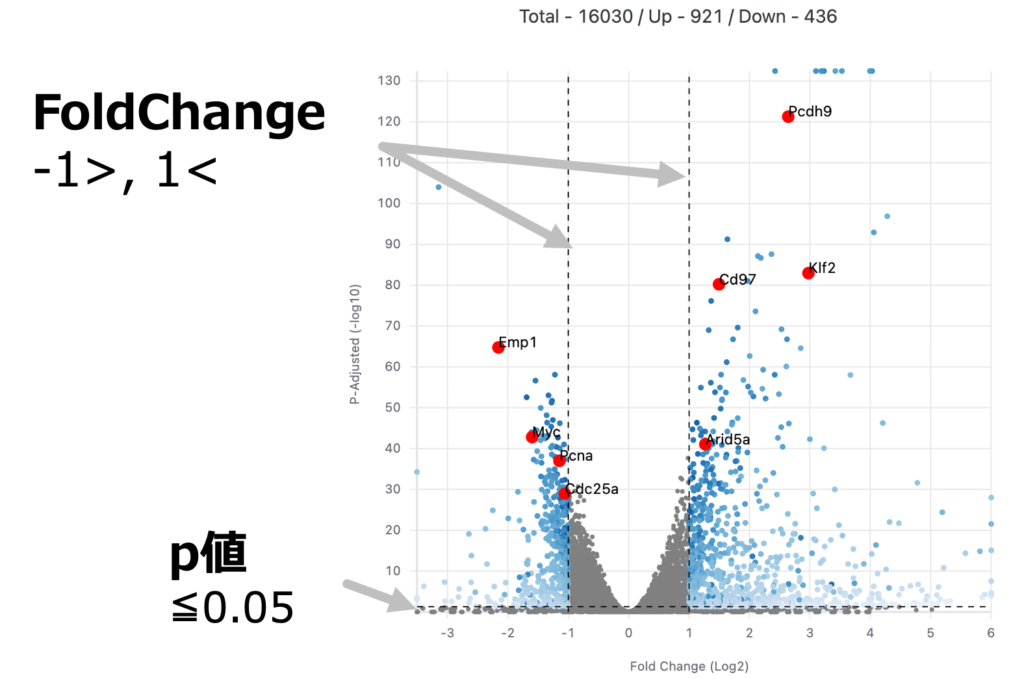
これを使って行うことは、基本的には一つです。それは、有意に差をもって変動している遺伝子を探すこと。どうするかというと、FoldChangeとp値で絞り込みを行います。
FoldChangeは、これは発現変動をlog2で表記したものです。2倍だと1、半分になると-1になります。発現変動が2倍以上、または半分になった遺伝子を絞り込む場合は、1以上、-1以下のものを絞ります。
もう一つは、p値と呼ばれる数字です。統計的な検定の結果で、一般的には0.05より小さいと統計的に有意をされます。統計的に有意でないと、変動している確らしさが低いため、排除することが目的です。
これらを組み合わせると、「統計的有意に、発現が倍、もしくは半分になった遺伝子」を抽出することができます。
ボルケーノプロット
これを図に表したものがボルケーノプロットです。横軸にFoldChange、縦軸にp値を取ります。火山噴火みたいだからボルケーノプロットと呼ぶそうです。
図の縦に2本ある点線がFoldChange「-1」と「1」、X軸すれすれに見えるのがp値が0.05の線です。先ほどの絞り込み作業は、この青いドットのエリアを探す作業だったというわけです。
エクセルで結果を納品されている場合は、エクセルでもなんとか図を作ることができます。あるいは、Rという統計処理に特化した言語があるので、それで描くことも可能です。(具体的な方法はタカラバイオさんのウェブサイトに動画で公開されています。)
エクセルの場合は、絞り込む→ブロット作成を逐次繰り返していくことになります。
インタラクティブレポートなら
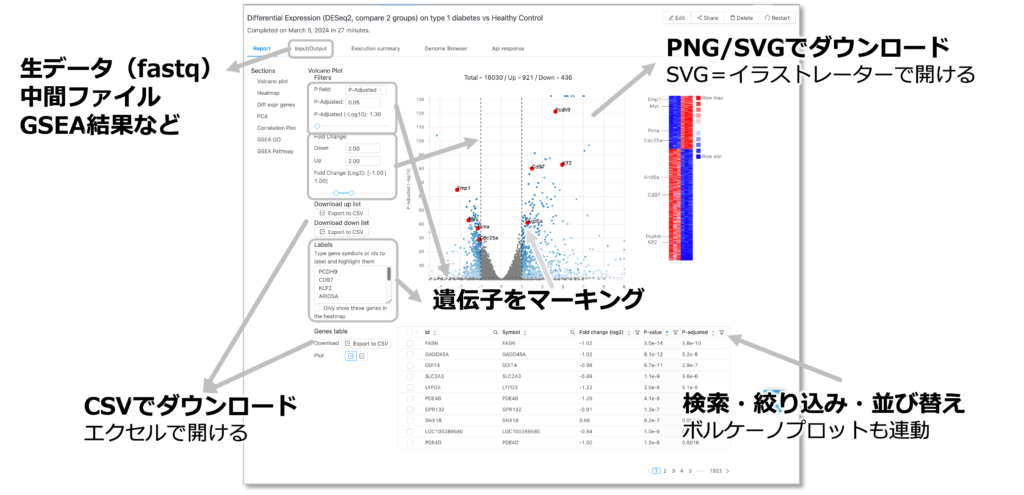
インタラクティブレポートは、この作業をブラウザ上でGUI(グラフィカルユーザーインターフェース)で行うことができます。ほとんどはマウスで操作可能です。
中央にボルケーノプロットがあり、左にはFoldChangeとp値の操作パネルがあります。変更はリアルタイムにボルケーノプロットに反映されます。
また、ボルケーノプロットの下には元となっている遺伝子のリストがあり、こちらでも絞り込みを行うことができます。検索、フィルター、並び替えが可能です。
ボルケーノプロットには、遺伝子のラベルをつけることも可能です。
これらは、右のクラスタリング・ヒートマップとも連動しています。
プロットはPNGというイメージファイル、SVGというベクターファイルでダウンロードすることができます。
SVGはAdobeのイラストレーターで開くことができるので、文字の変更や色の変更なども可能です。
絞り込まれた結果は、CSVでダウンロードすることができます。
エクセルで開けるのは同じですが、すでに絞り込まれているので、数万行という大きなファイルと格闘することはありません。
レポートに含まれるその他のデータ
インタラクティブレポートには、その他にも一般的な標準解析に含まれるデータが格納されています。
QC、アライメントの結果やゲノムブラウザなども搭載しています。PCAや相関プロットもあるので、サンプル群の中に外れ値があればそれを見つけることができます。エンリッチメント解析(GSEA)GOやpathwayの解析も含まれています。また、input/outputタブでは、生データ(fastq)やBAMなど中間ファイルなどもダウンロード可能です。
再計算要検討の例
時折、再解析を検討した方がよい場合があるので、QC、アライメント、PCA・相関プロットはぜひ一度は確認してほしい内容です。
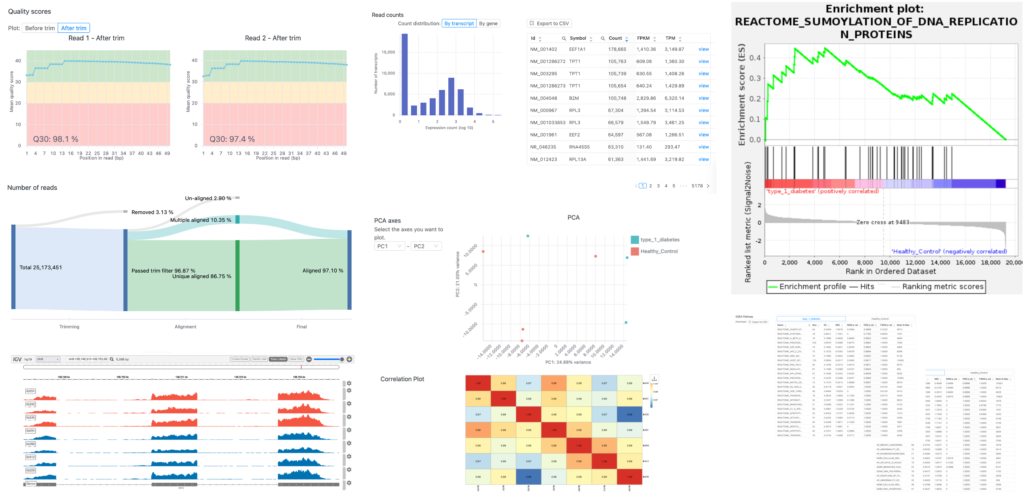
PCA・相関プロット
相関プロットは、縦軸・横軸に同じものを並べて、どのくらい同じかを可視化したものになります。例示したの相関プロットでは、左上から右下に赤いマスがありますが、これらは自分と自分を比べているところなので、100%一致です。青いマスがあります。これはもしかしたら、他のサンプルと比べてデータが違っている可能性があります。
いわゆる外れ値です。外れ値がある場合は、そのサンプルを除いて、再度比較解析をするとよりよい結果になる可能性があります。
PCAは主成分分析で、サンプル同士の似ている度合いを数値化しプロットしたものです。似ていれば近くにプロットされます。同じ群のサンプルであるにもかかわらず、一つ離れてプロットされていれば、外れ値である可能性が高いと言えます。
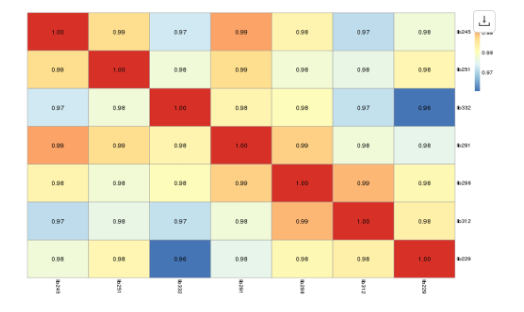
データQC
左から、リードの3’末端から5’末端になるように並べられています。緑のエリアは十分綺麗なデータですが、黄色、赤となるにしたがってデータの信頼度が低いことを示しています。この場合は、トリミングをやり直し方が良いかもしれません。
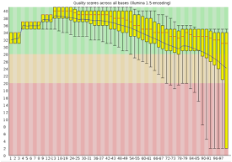
アライメント
左端がインプットのリード数で、右端がアライメントの状況を示しています。インプットされたリードが全部アライメントされれれば、両端の高さは同じになります。
この図の場合は、40%くらいしかアライメントされていません。つまり60%くらいのリードは捨ててしまうことになります。もしかしたら、誤ったゲノム情報を参照した、という初歩的なミスかもしれません。
いずれにしても、アライメントをやり直した方がよいかもしれません。
比較のやり直しはカウントデータがあれば可能です。ですが、トリミングとアライメントはfastqに戻ってやり直しになります。
再解析
典型的な受託会社の場合
多くの場合は、pdfやエクセルでレポートが納品されます。この場合、先ほどの再解析を行おうとするとどうなるでしょう。
比較のやり直しは、追加解析ということで追加料金を払えばできそうです。ただ、記載がない受託会社もあるので、問い合わせが必要です。
fastqに戻ったやり直しは、どこも記載がありません。とはいえ、ほぼ解析やり直しですから、元の料金と同じくらいはかかるのではないか?という気がします。こちらも問い合わせするしかありません。
解析をどう行っているかにかかわらず、内容を確認し、修正を依頼し、再度内容を確認して…という繰り返し作業は避けられません。
受託会社の場合、多くの場合、再解析など価格が公開されていません。当然「問い合わせ」が必要になります。問い合わせ、見積もり、発注、これで何日かロスすることになります。早くても2〜3日はかかるのではないでしょうか?
インタラクティブレポートの場合

インタラクティブレポートは単なるレポートではなく、解析機能が含まれます。また、fastqも格納しているので、fastqに戻って再解析も可能です。
これらは、追加費用なく何度でも行うことができます。比較の再解析・追加解析は、最速30分で完了します。再解析でfastqからスタートしても1時間程度で完了します。並列処理していますので、サンプル数がいくらあっても、処理時間は大きく変わりません。
追加料金もありません。
インタラクティブレポートなら、やり取りや問い合わせの待ち時間の間に、解析は完了します。
バイオロジストの本分はバイオロジー
料理で例えると、NGSのデータ解析は、素材加工に当たります。皮を剥いたり、カットしたりして料理ができるようにする「仕込み」の段階です。
バイオロジストは料理の部分にもっと時間が使えるようになるべきです。つまり、目的の遺伝子を絞り込んだ後がバイオロジストの本来の仕事です。
その遺伝子が何をしているのか?他に影響を与えていることはないか?生物学的な意義は何か?
NGSのデータを解析することそのものではないはずです。
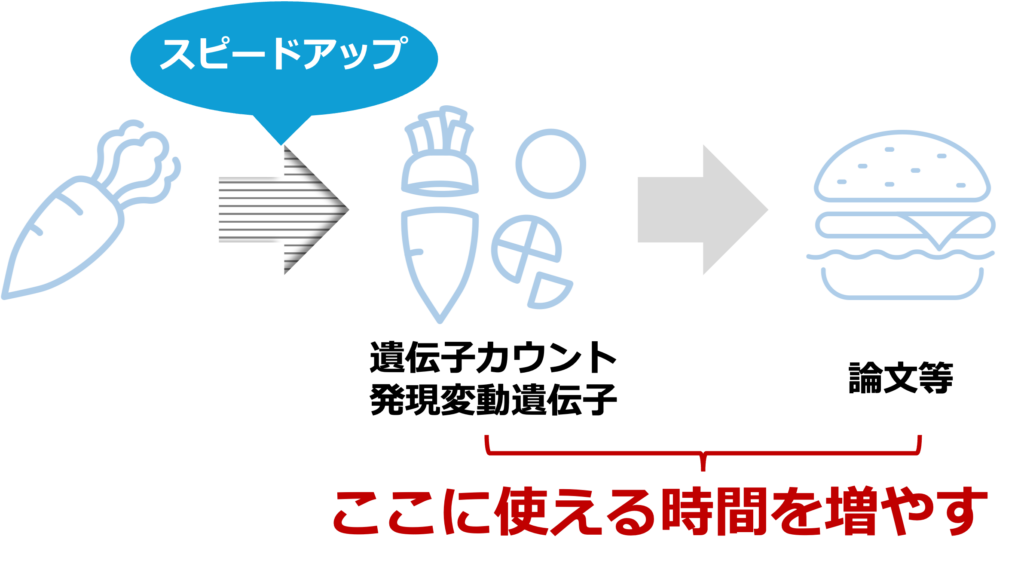
インタラクティブレポートで、スピードアップが可能です。大きいエクセルと格闘したり、あらたにRを覚えたり、誰かにお願いしたりする必要はありません。
インタラクティブレポートのメリット
- インタラクティブレポートでスムーズな解析:エクセルやRと格闘する必要はありません。
- グラフやデータはダウンロード可能:SVGはイラストレーターで開くことができます。
- 追加解析・再解析も制限なし:生データ(fastq)からやり直しても1時間程度です。
これまでRNA-Seqのデータ解析は、大変、時間がかかる、と思っていませんか?むしろそれが常識になっているかもしれません。
インタラクティブレポートはRNA-Seqのデータ解析のスピードアップが可能です。