
A Deep Dive Into Differential Expression
細胞の機能は、どの遺伝子が活性化されているかによって主に決定されます。 したがって、条件間で差次的に発現される遺伝子を見つけることは、多くの場合、疾患組織と正常組織、または未処理サンプルと処理サンプルの間の表現型変化の分子機構を理解するために必要な部分です[1]。 RNA-seq は、細胞の転写状態を定量的に読み取ります。 生のカウントデータから、一般に示差発現と呼ばれる分析で、サンプルのグループ間の統計的に有意な差を決定できます。 Basepair は、このような分析を実行できるようにするいくつかのパイプラインを提供します。この投稿では、分析の設定と結果の解釈について詳しく説明します。
分析のセットアップ
発現差分析を実行する前に、「発現カウント (STAR)」パイプラインを通じてサンプルを分析する必要があります。 発現差解析にはさまざまなツールが公開されていますが、Basepair は非常に引用されている DESeq2 ツールを使用しています [1、2]。
次のステップは、実験計画を検討することです。 最も単純なセットアップでは、比較するサンプルの 2 つのグループがあります。 ただし、Basepair は、(1) 3 つ以上のグループを比較して各グループの固有の遺伝子を取得するか、ANOVA 分析を実行する、(2) 時間経過実験、および (3) ネストされた条件など、より複雑なデザインに対応できます。 ANOVA を使用すると、3 つ以上のサンプル グループを比較し、 差異の有意性を示す1 つの p 値 (遺伝子ごと) の グループ間 を取得できます。 パイプラインは各グループに 1 つのサンプルを収容できますが、 理想的にはグループごとに少なくとも 3 つのサンプルが必要であること に注意してください。
分析を実行する前に考慮すべきパラメーターが他にもいくつかあります。 サンプルにスパイクインを追加した場合、分析でそれを使用してサンプルを正規化し、結果を向上させることができます。 スパイクインは、より正確な発現定量を可能にするために、指定された量でサンプルに追加される短い既知の配列です。 考慮すべきもう 1 つのオプションは、遺伝子または転写産物に対して差次的発現を実行するかどうかです。
パラメーターを選択したら、分析を開始する準備が整いました。 次のセクションでは、分析結果の解釈について説明します。
結果の解釈
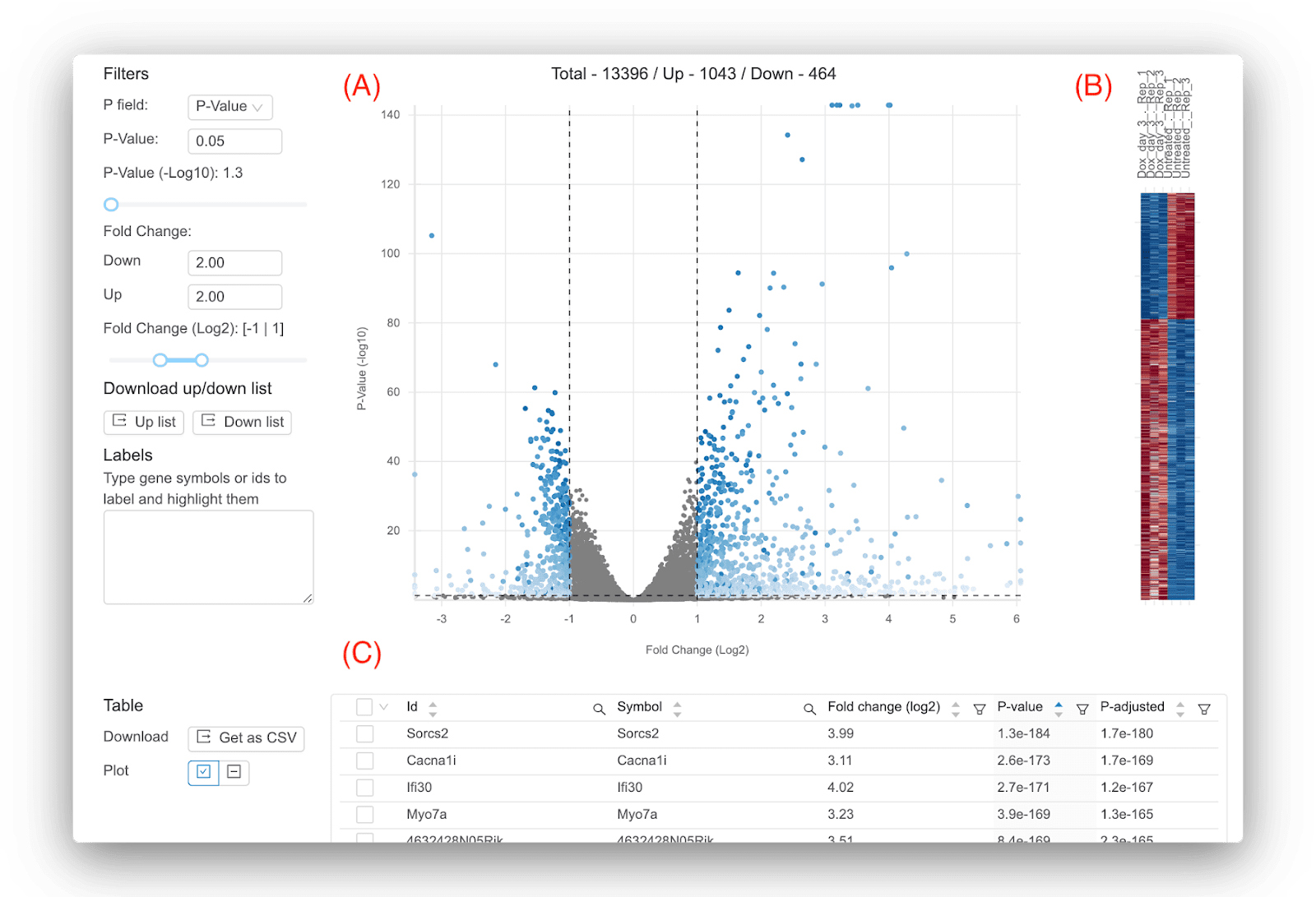
Basepair にはいくつかのインタラクティブな表と図が用意されているため、結果を調べてすぐに答えを得ることができます。 図 1 は、差分式の実行による主な結果を示しています。ボルケーノ プロット、ヒートマップ、および log2 倍数変化、p 値、調整済み p 値などの遺伝子ごとの統計を含むテーブルです。 デフォルトでは、図に表示される重要な遺伝子は、p 値 0.05 および log2 倍率変化の +/- 1 を使用して定義されますが、独自のしきい値を自由に選択できます。 に焦点を当てることです ただし、重要な遺伝子を定義するためのアドバイスの 1 つは、調整された p 値 。これにより、p 値を使用する場合と比較して偽陽性の結果が得られる可能性が低くなります。 調整された p 値のしきい値に真の「最適な」値はありませんが、一般的に使用される値は 0.01 または 0.05 です。
二次分析
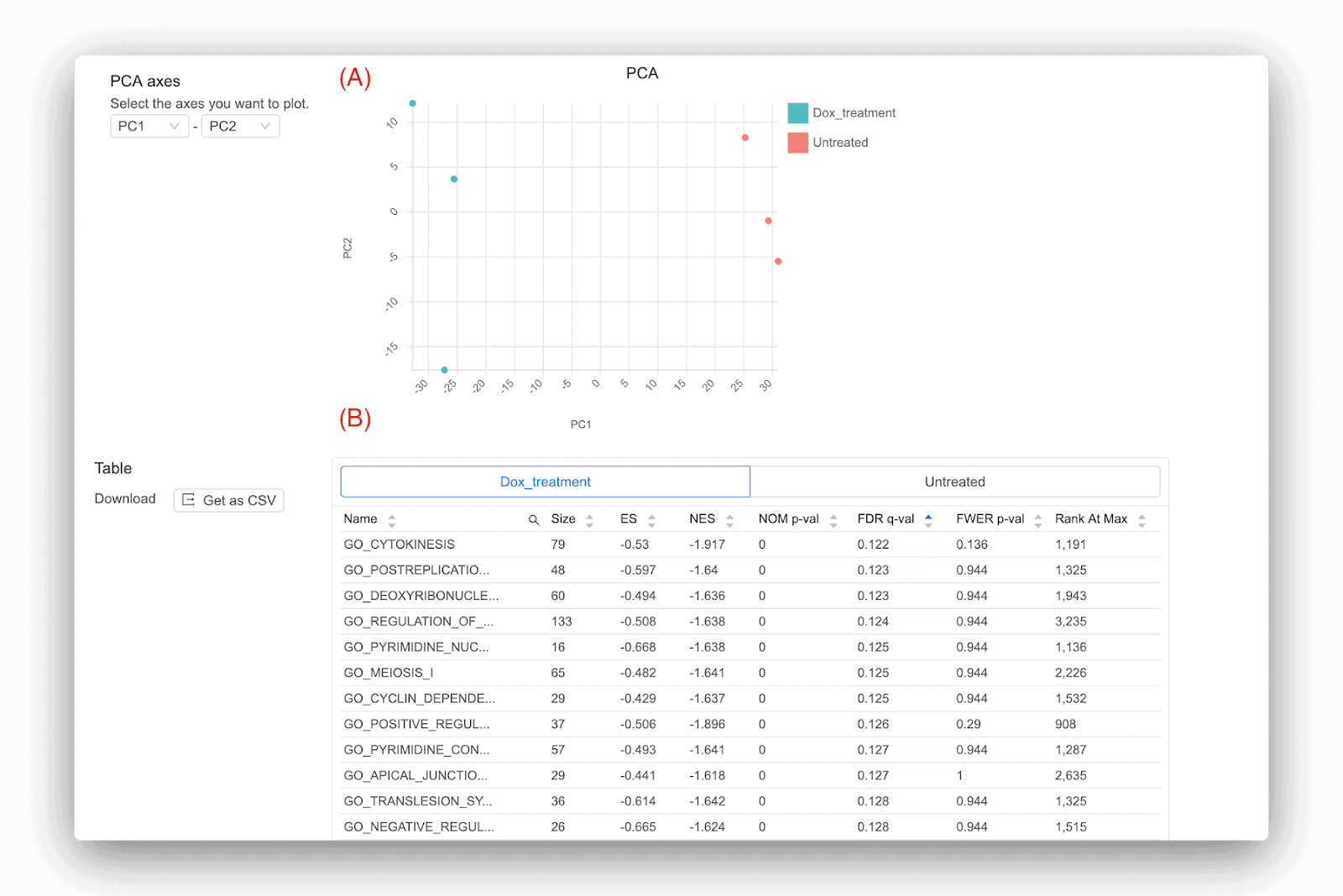
Basepair は、発現差に加えて、いくつかの二次解析も提供します (図 2)。 1 つは主成分分析 (PCA) で、サンプルがどのようにグループ化されるかを示す次元削減アルゴリズムです。 PCA は発現したすべての遺伝子を分析に使用し、次のようなさまざまな方法で役立ちます。(1) サンプルが予想どおりにクラスター化しているかどうかを明らかにするのに役立ちます。(2) サンプルのいずれかが誤ってラベルされているかどうかを明らかにします。または、(3) を明らかにします。異常値がある場合。 Basepair の優れた点の 1 つは、結果にそのような問題が見つかった場合、これらの問題を考慮しながら別の分析を実行できることです。 外れ値を削除したり、任意のサンプルのラベルを付け直したりすることができます。そうしないと結果が歪められ、得られる重要な遺伝子が大幅に少なくなる可能性があるためです。
Basepair が提供するもう 1 つの分析はパスウェイ分析です。 これは、サンプル内で発生する生物学的変化をより詳細に把握するための優れたツールです。 Basepair は、広く使用されている Gene Set Enrichment Analysis (GSEA) ツールを使用します [3]。 GSEA は、遺伝子セット (KEGG 経路など) のデータベースを使用し、遺伝子セット内の遺伝子が偶然に予想されるよりも上方制御または下方制御される可能性が高いかどうかを確認する統計的検定を実行します。 したがって、GSEA は、サンプル グループのいずれかでどの遺伝子セットが豊富であるかを示します。 Basepair によって得られる代表的な結果の一部を図 2 (B) に示します。 各遺伝子セットについていくつかの統計が報告されていますが、注目すべき主な統計は NOM p-val と FDR q-val です。 後者は誤検知の結果が得られる可能性が低いため、優先する必要があります。 FDR q-val に使用される一般的なしきい値は、0.01、0.05、0.1、さらには 0.25 です。
References
1. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. BioMed Central Ltd; 2014; 15(12):550.
2. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics; 2013 Mar 9; 14(1):1-1.
3. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005 Oct 25; 102(43):15545–50.