
主な特徴
この度、BasepairのシングルセルRNA-seq(scRNA-seq)パイプラインが大幅にアップデートされました。新しく機能を追加するとともに、効率性、使いやすさの面で多くの改善を行いました。
1. Seuratバージョン3
- Basepairのパイプラインは現在、Seuratバージョン3とその改良点の多くを活用しました。
2. より速く、よりインタラクティブなレポート
- PCAとt-SNEに加えて、UMAPによるクラスタリングがレポートに表示されるようになりました。
- 細胞間での発現を可視化するために、遺伝子を素早く検索できます。
- 発現変動解析の結果の表をソートおよびフィルタリングできるようになりました。「Action」 列の下にある虫眼鏡アイコンをクリックして可視化できます。
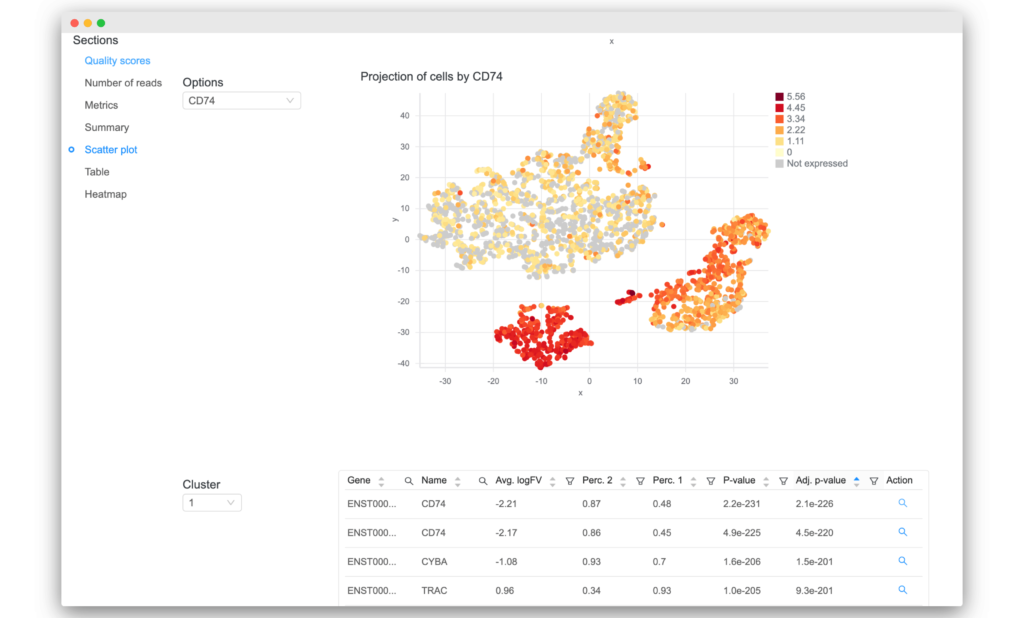
3. データ品質レポートの改善
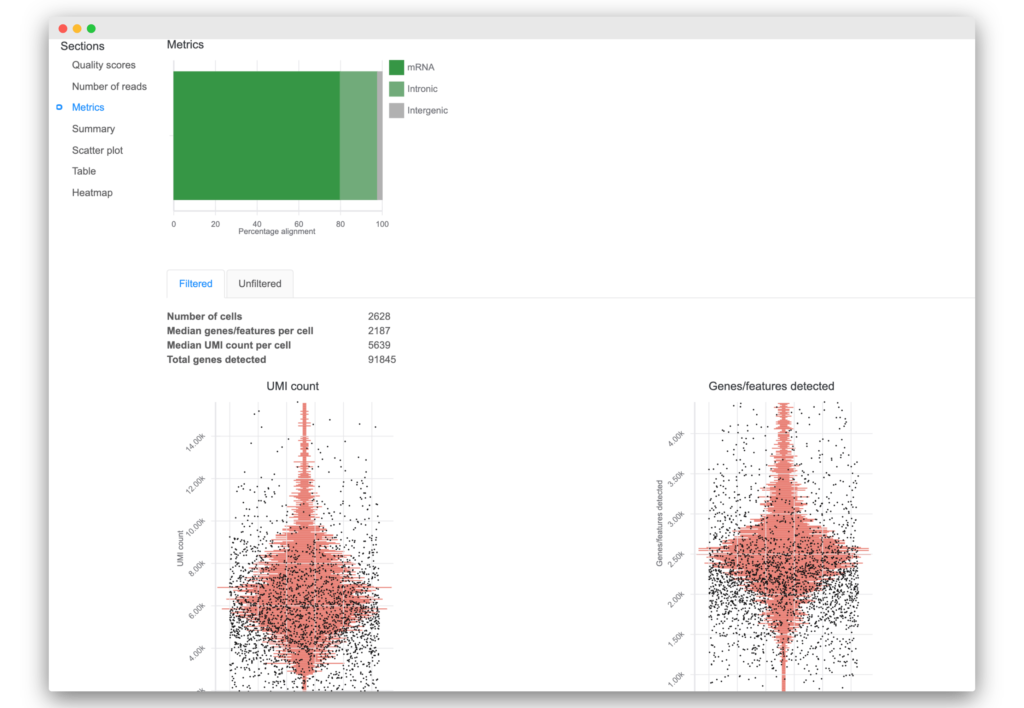
- リードの品質とマッピングメトリクスの視覚化が更新されたことにより、迅速に結果を検証することができます。
- 細胞数、検出された総遺伝子数、細胞あたりの UMI カウントの中央値などの追加レポート統計情報が追加されました。
- 各細胞の主要な品質メトリクスの新しい視覚化にアクセスできます:
- UMI カウント
- 検出された遺伝子/フィーチャー
- ミトコンドリアの割合(利用可能な場合)
検証
一般に公開されているデータセットを用いて、Basepairのパイプラインの有効性を実証しました。検証には、10X Genomics社の末梢血単核球(PBMC)データセットを使用しました。このデータセットはSeuratの著者によって以前に解析されており、その結果はオンライン上で公開されています[2]。我々の更新したscRNA-seqパイプラインのステップを図1に示します。
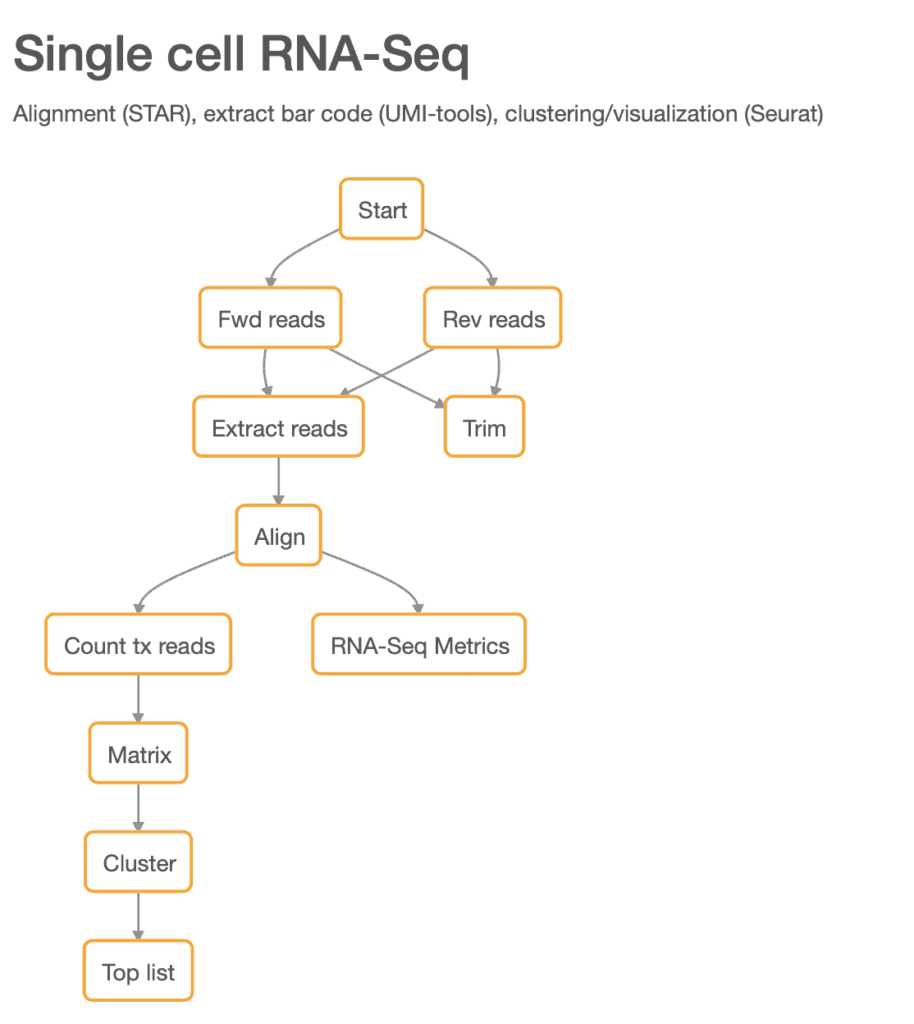
図1:新しいscRNA-seqパイプライン。
scRNA-seq解析における重要な解析の一つは、細胞がどのようにクラスター化しているかを確認することです。これは一般的にt-SNEまたはUMAPアルゴリズムで行われます。図2は、BasepairのパイプラインによるUMAPの結果とSeuratの著者によって報告された結果を比較したものです。全体として、クラスターの数と互いの相対的な位置は基本的に同じです。なお、彼らのデータは10x Genomicsワークフローを使って前処理されているため、若干の違いがあります。
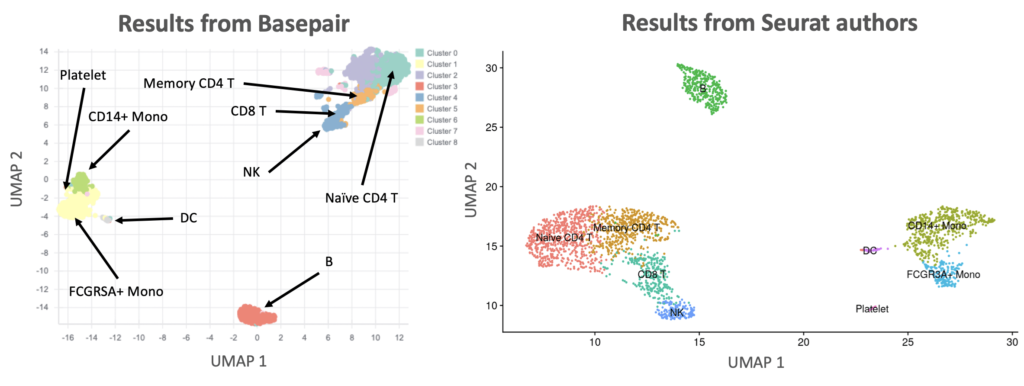
図2:UMAPの結果の比較。左:Basepairの結果。右: Seurat authorの結果。どちらのプロットも、同定された細胞タイプを表示しています。
scRNA-seqで一般的に行われる2つの追加解析は、(1)グループ間の発現に変動のある遺伝子と、(2)細胞全体にわたる遺伝子の発現パターンの可視化です。Seuratの著者らは、クラスター間で差次的に発現している9つの遺伝子をプロットしました(図3)。各遺伝子の発現が特定の細胞群に限定されていることがわかります。我々も同様の解析を行い、同じ9つの遺伝子の発現パターンをプロットしました(図4)。この2つの図には類似性が見られます。
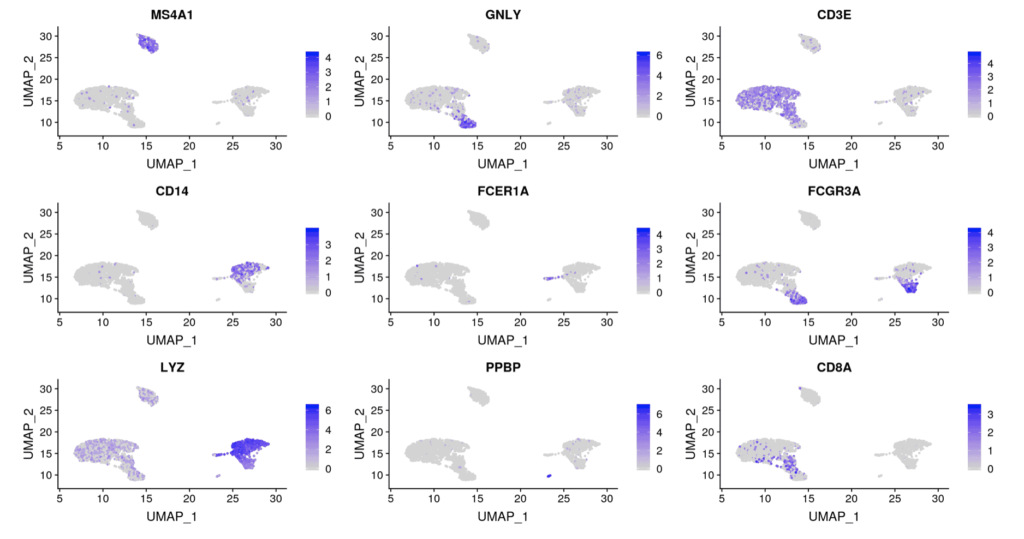
図3:Seurat著者による結果。Seurat著者による解析で同定された9つの差次的発現遺伝子の発現値のプロット。
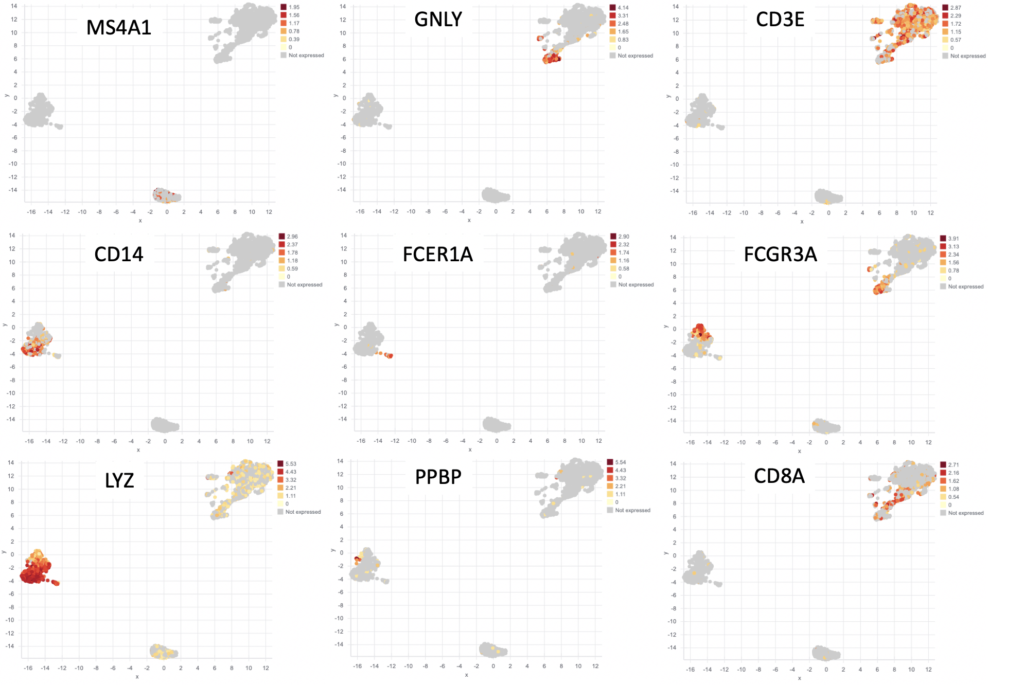
図4:Basepairの結果。図3と同じ9遺伝子ですが、Basepairの解析データを用いてプロットしました。
今後の課題
Seuratバージョン3には、独立したscRNA-seqデータセットをマージするエキサイティングな新ツールも搭載されました。現在、この機能を利用するためのパイプラインをBasepairに追加する作業を行っています。ご期待ください!
それまでの間、iwai-chem.basepairtech.comにアクセスして、アップデートされたシングルセルRNA-seqパイプラインをお試しください。
参考文献
最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。