
アダプターや低品質塩基のトリミングは、シーケンスデータの解析パイプラインの重要な部分です。通常、RNAサンプルを分離して断片化した後、シーケンシングに必要な配列の末端にアダプターが付加されます(シーケンシングの技術的背景については[1,2]を参照してください)。これらのアダプターは、下流の解析で処理する前にシーケンスされたリードから除去する必要があります。さらに、低品質塩基の除去も必要です。リード内の各塩基にはQ値が割り当てられ、これはその塩基が誤ってコールされた確率の負の対数として定義されます。Illuminaの特異的なシーケンスケミストリにより、Q値はリードの3’末端に向かって低下する(品質が悪くなる)傾向があります。これらの低品質領域は、マッピングや変異コールなどのダウンストリーム解析に悪影響を及ぼす可能性があります。
しかし、RNA-seqにおけるクオリティトリミングの有用性については議論があります([3,4,5]を参照してください)。これまでの研究を要約すると、RNA-seqにおけるトリミングは一般的にリード数の減少につながるが、マッピング可能なリードの割合は増加します。しかし、これらの研究者たちは、積極的なトリミングは、発現変動解析や発現推定などのダウンストリーム解析に悪影響を及ぼす可能性があることを示唆しています。また、高品質のRNA-seqデータセットを持っている場合、クオリティトリミングはほとんど影響を与えないので、必要ないかもしれないことに注意すべきです。質の低いデータセットでは、質の高いトリミングが最も大きな違いをもたらします。 したがって、これらの点をさらにユーザーに示すために、あるRNA-seqサンプルでの解析例を示します。
データ例
151bpのペアエンドリードが約2300万本ある単一のRNA-seqサンプルを使用します。リード内の位置によるQ値の分布を図1に示します。Illuminaのデータであるため、Q値は3’末端に向かって減少しています。リードをSTARでマッピングした結果、約80%のリードがユニークまたはマルチマッピングされました(図2)。
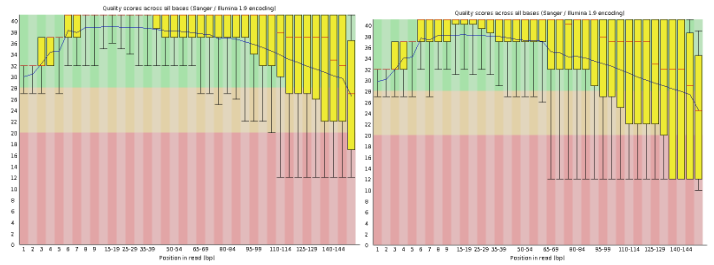
図1:フォワードリードとリバースリードのペアにおける位置ごとのQ値分布。
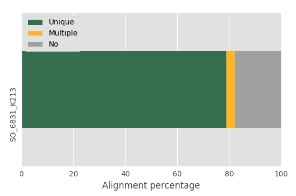
図2:一意にマップされたリード、マルチマップされたリード、マップされていないリードの割合。
次に、複数の異なるQ値閾値(5,10, …, 30)を用いて、生のリードをトリミングしました(fastp trimmerを使用)。Fastpは、リード上のウィンドウの平均Q値を調べ、その平均が指定されたQ値のしきい値を下回った場合にトリミングします。図3と図4は、Qのしきい値を増加させながら、一意にマップされたリード、マルチマップされたリード、マップされていないリードの割合とカウントをそれぞれ示しています。この結果から明らかになったことは、トリミングによってリードの総数は減少するが、一意にマップされたリードの割合と総数は増加するということです。
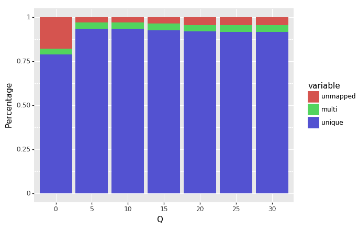
図3:Q閾値と、一意にマップされたリード、マルチマップされたリード、マップされなかったリードの割合の関係。
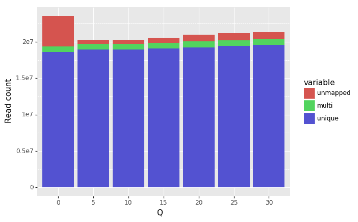
図4: Qの閾値と、一意にマップされたリード、マルチマップされたリード、マップされなかったリードの数の関係。注:Q値が高くなるにつれてリードの総数が増えるのは(Q=5以降)、fastpにはデフォルトで、40%以上の塩基が別の品質しきい値以下のリードを除去するオプションがあるためです。そのため、Qが高くなるにつれて低品質のリード領域が除去されるため、全体として除去されるリードは少なくなります。
最後に、結果に機能的な意味を持たせるために、各データセットの遺伝子発現レベルを推定しました。未トリミングデータと各トリミングデータセット間の発現値の相関を計算しました(図5)。すべての相関が非常に高いことがわかったが、より積極的なトリミング(Qしきい値20あたりから)により、相関は低下し始めました。Williams et al. (2016) も似たようなことを発見しましたが、さらに踏み込んで、RNA-seqサンプルと同じサンプルのマイクロアレイデータを相関させました[3]。彼らもまた、積極的なトリミングによって相関が著しく低下することを発見しました。
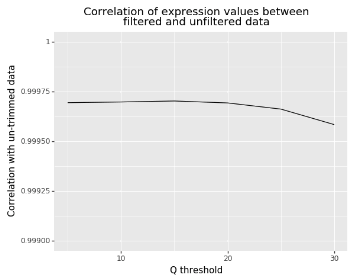
図5:未トリミングデータとQ閾値を変えたデータとの遺伝子発現の相関。
結論
我々の1つのRNA-seqサンプルで得られた知見は、概して先行研究と一致していします。すなわち
- Qualityトリミングはリード全体の数を減少させるが、ユニークにマップされたリードの合計と割合は増加させます。したがって、ダウンストリーム解析により有用なデータが得られます。
- あまりにアグレッシブなQuality Trimmingは、ダウンストリーム解析(この例では遺伝子発現の推定)に悪影響を与える可能性があります。
- 従って、RNA-seqデータに軽いトリミングを取り入れることは、私たち自身の知見や研究コミュニティの知見から動機づけられています(私たちはQ閾値10を使用しています)。
参考文献
1. Shendure, J., & Ji, H. (2008). Next-generation DNA sequencing. Nature Biotechnology, 26(10), 1135–1145. http://doi.org/10.1038/nbt1486
2. Ozsolak, F., & Milos, P. M. (2011). RNA sequencing: advances, challenges and opportunities. Nature Reviews Genetics, 12(2), 87–98. http://doi.org/10.1038/nrg2934
3. Williams, C. R., Baccarella, A., Parrish, J. Z., & Kim, C. C. (2016). Trimming of sequence reads alters RNA- Seq gene expression estimates. BMC Bioinformatics, 1–13. http://doi.org/10.1186/s12859-016-0956-2
4. MacManes, M. D. (2014). On the optimal trimming of high-throughput mRNA sequence data. Frontiers in Genetics, 5, 13. http://doi.org/10.3389/fgene.2014.00013
5. Del Fabbro, C., Scalabrin, S., Morgante, M., & Giorgi, F. M. (2013). An extensive evaluation of read trimming effects on Illumina NGS data analysis. PLoS ONE, 8(12), e85024. http://doi.org/10.1371/journal.pone.0085024
最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。