
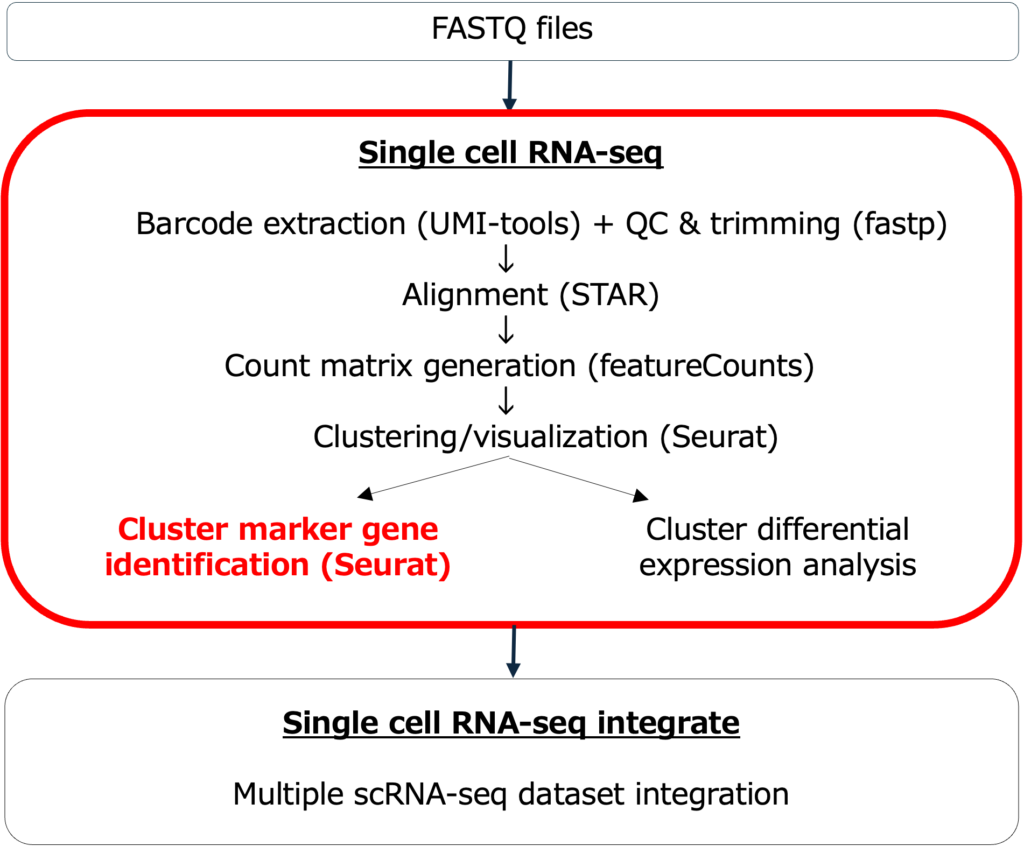
背景
シングルセル解析では多くの場合、クラスタリング解析を行います。そこでは、クラスター内のマーカー遺伝子を同定することが重要です。これにより、サンプル内の各クラスターの細胞種を特定することができます。
Seuratによるマーカー遺伝子の同定は、Basepairの「Single cell RNA-seq」「Single cell RNA-seq integrate」パイプラインに組み込まれています。Seuratによるマーカー遺伝子の同定が自動的に実行されます。パイプラインの実行は数回クリックするだけで、とても簡単です。
Basepairでのマーカー同定解析の種類
Basepairでは3種類のマーカー同定解析が可能です。1サンプルの場合は、クラスターマーカー遺伝子のみが実行されます。一方で、「integrate」パイプラインでは、すべての解析が実行されます。
1. クラスターマーカー遺伝子(Cluster marker genes)の同定
選択したサンプルのすべてのクラスターに対して、各クラスター内の遺伝子の発現変動遺伝子解析を実行します(図2)。クラスターマーカーの可能性のある遺伝子は 「FindAllMarkers」で同定されます。この解析は、1つのサンプルグループを評価するのに便利です。
2. 保存マーカー遺伝子(Conserved marker genes)の同定
まず各サンプル内のクラスターで遺伝子発現の差分解析を行います。次に、サンプル間(例えば、トリートメントグループとコントロールグループ)で比較を行います。サンプルグループ間でクラスターに保存されている遺伝子を 「FindConservedMarkers」機能で同定します。この解析は、異なるサンプルグループ間で保存されている遺伝子マーカーを見つけるのに有用です。
3. 条件間で差のあるマーカー遺伝子(Markers differential between conditions)の同定
まず各サンプル内のクラスターで遺伝子発現の差分解析を行います。次に、異なるグループ・条件(例えば、トリートメントグループとコントロールグループ)間で比較します。サンプルグループ・条件間で発現変動している遺伝子が同定されます。この解析は、サンプルグループ・条件間のクラスターの違いを見つけるのに有用です。
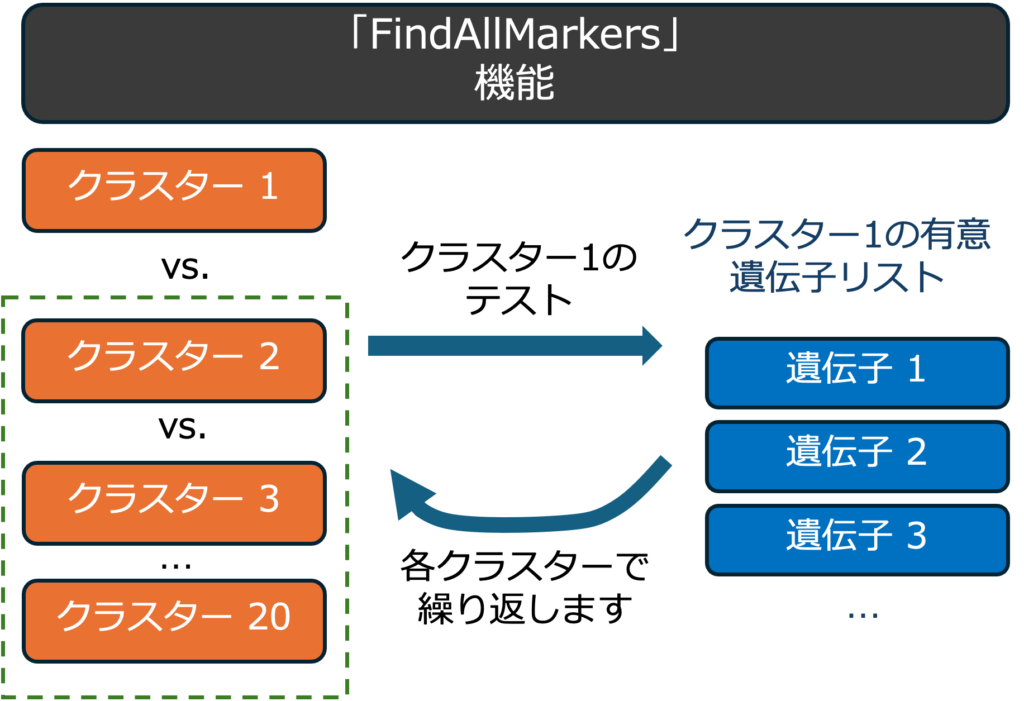
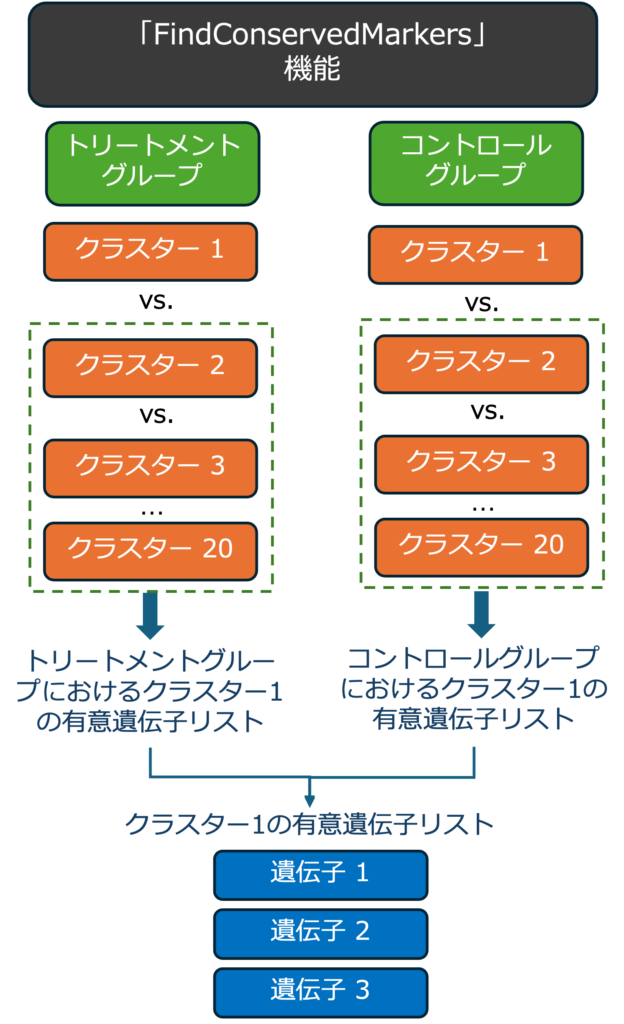
図4. 条件間で差のあるマーカー遺伝子同定のワークフロー((1)からの改変)。
結果
「Report」タブでは、同定されたクラスター遺伝子マーカー、保存遺伝子マーカー、条件間で差のある遺伝子マーカーのリストがすべて表として表示されます。サンプル間のクラスターで同定されたすべてのマーカー遺伝子の発現を示すヒートマップも表示されます。
アウトプットテーブル
結果を示すテーブルは、タブによってクラスターマーカー遺伝子、保存マーカー遺伝子、条件間で差のあるマーカー遺伝子の情報を切り替えて見ることができます。アウトプット表はCSVファイル形式でエクスポートでき、下流の解析に利用できます。
ドロップダウンでクラスター番号を選択して、クラスターを切り替えることができます。また、「Sample」ドロップダウンで、をクリック、異なるサンプルの結果を見ることができます。
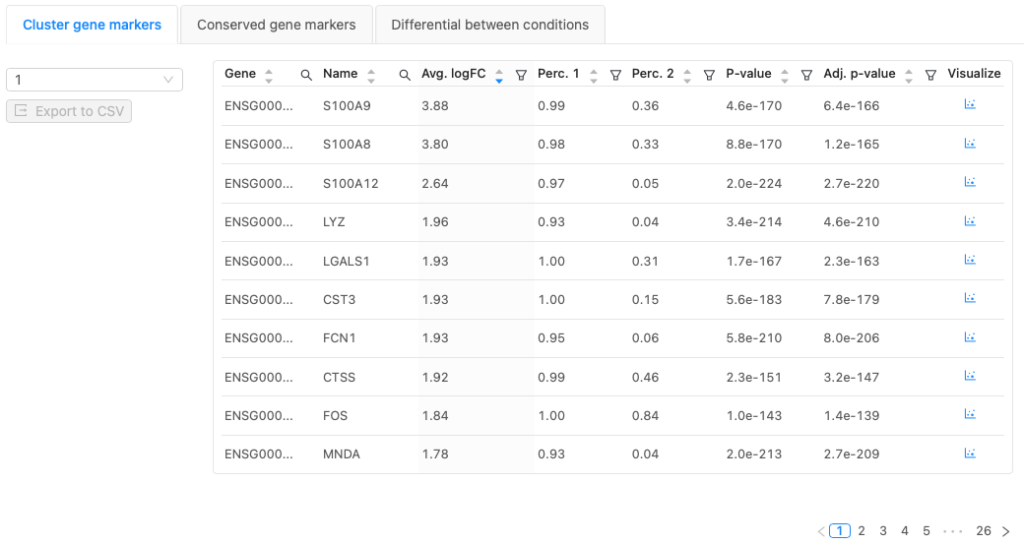
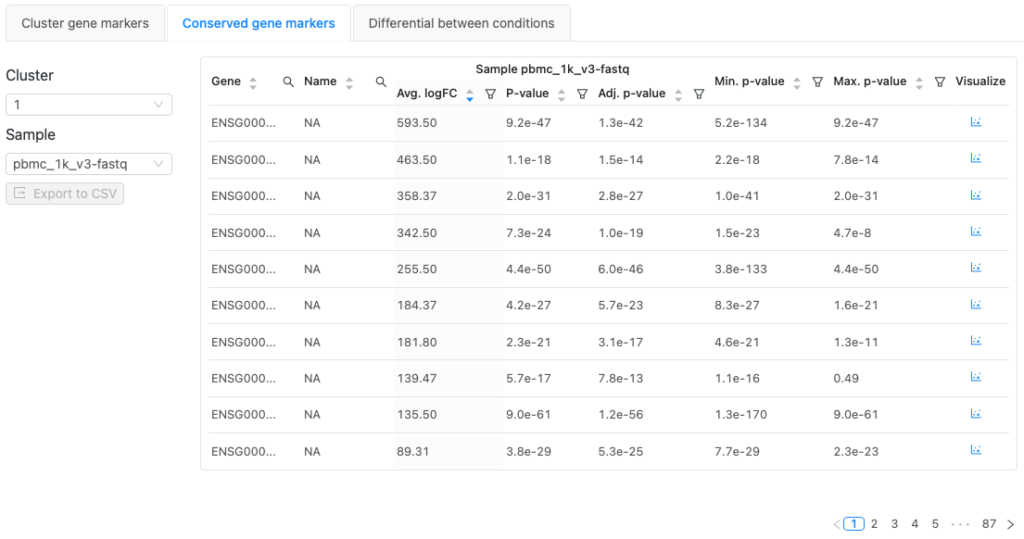
テーブルの項目
| Gene | 遺伝子記号 |
| Name | 遺伝子名 |
| Avg. logFC | サンプル・グループの平均「log2 fold change」発現量。値が大きいほど、その遺伝子はクラスター内で高発現しています |
| Perc. 1 | あるグループのクラスターにおいて、その遺伝子が検出された細胞の割合Perc. 2: あるサンプルについて、その遺伝子が他のクラスターで検出された細胞の割合(平均) |
| P-value | グループについて多重検定補正をしていないp値 |
| Adjusted p-value | データセット中の全遺伝子を用いたBonferroni補正に基づき、有意性を決定するために使用されるグループの調整p値 |
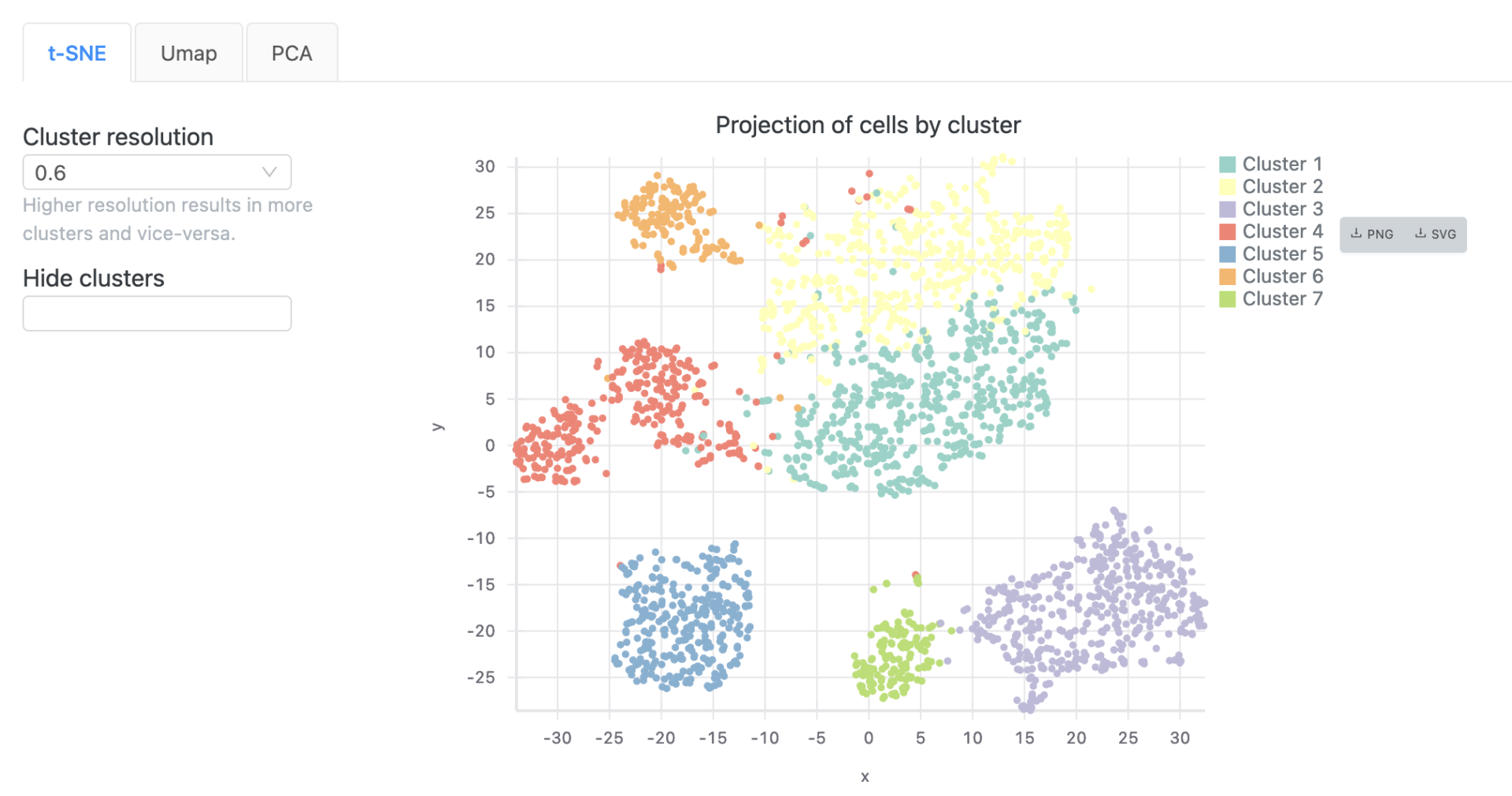
| Visualize | 散布図(t-SNE、UMAP、PCA)およびバイオリンプロットで、選択した遺伝子の全クラスタにおける遺伝子発現レベルの分布を可視化することができます。 |
テーブルとプロットとの連動
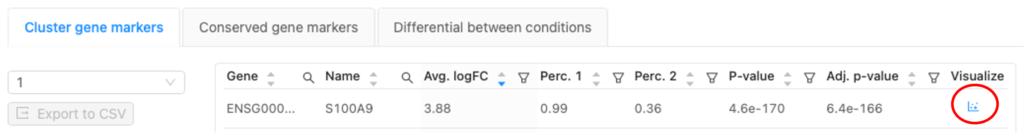
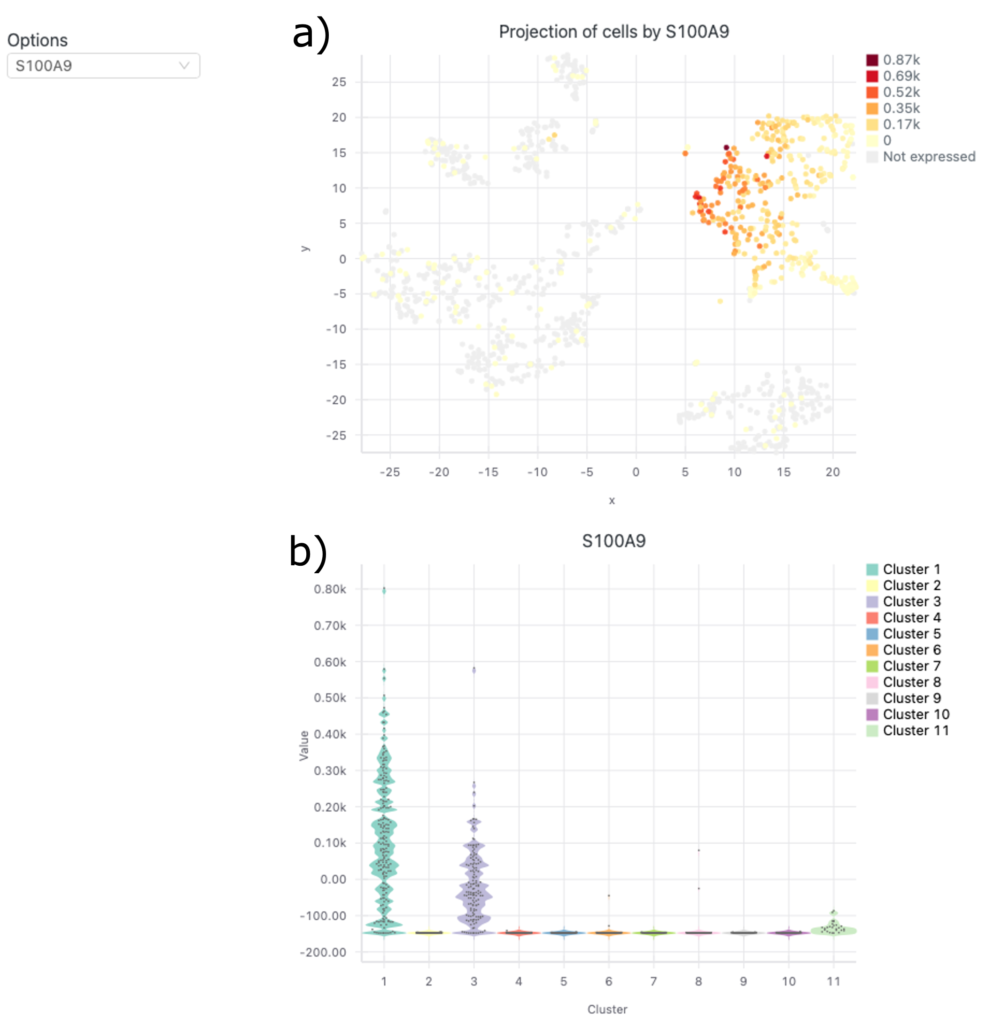
アウトプット表(図7の赤丸)に表示された遺伝子の「Visualize」ボタンをクリックすると、全クラスタにおけるその遺伝子の発現レベルの分布を散布図(図8a)とバイオリンプロット(図8b)として見ることができます。例えば、下の例では、S100A9のVisualizeをクリックした際のクラスター表示です。
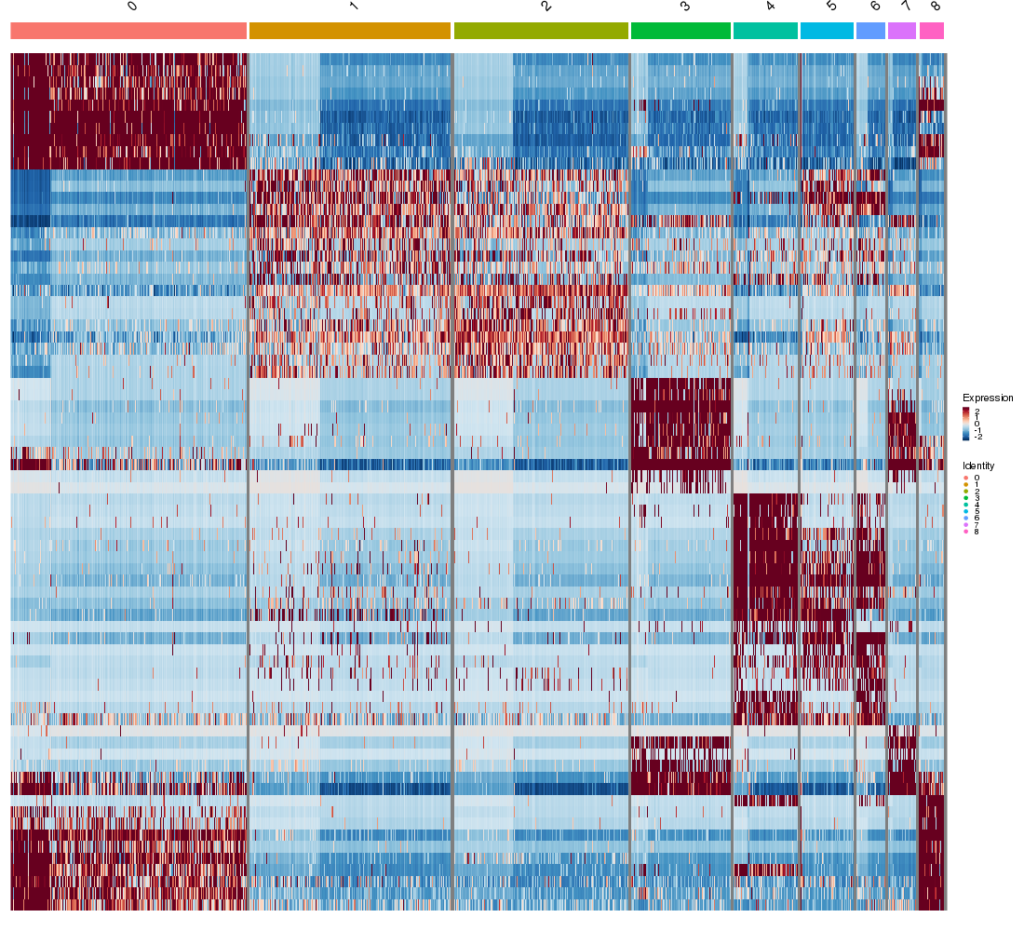
ヒートマップ
散布図やバイオリンプロットと同様に、サンプル間の全クラスターで同定されたマーカー遺伝子の発現もヒートマップで示すことができます(図9)。それぞれのクラスターに属する細胞を列で示し、遺伝子を行で示しています。凡例の「expression」は遺伝子発現レベル、「Identity」はクラスター番号を示しています。例えば、ヒートマップのクラスター0は散布図のクラスター1に相当します。
図9の各交点における色の濃さは、細胞における遺伝子発現のレベルを示します。あるクラスターで一貫して赤い遺伝子は、そのクラスターで高発現していることを示します。一方、あるクラスターで一貫して青い遺伝子は、そのクラスターで低発現であることを示します。
アウトプットファイル
「Input/output」タブに、toplist zip(図10の赤枠)フォルダが生成されます。すべてのマーカー遺伝子リストとヒートマップの結果を含みます。これらの詳細なマーカー遺伝子リストとヒートマップは、論文やさらに下流の解析に使用することができます。

Toplistのzipフォルダには以下のファイルが含まれています:
- 0.1-2.0の解像度の上位10遺伝子のヒートマップ(png形式)
- (例:heatmap_image_top10_RNA_snn_res.0.1.png)
- 解像度0.1-2.0の上位10遺伝子のヒートマップ(json形式)
- (例:heatmap_top10_RNA_snn_res.0.1.json)
- 解像度0.1-2.0のマーカー遺伝子のリスト(json形式)
- (例:markers_RNA_snn_res.0.1.json)
- 解像度0.1-2.0の上位10個のマーカー遺伝子のリスト(json形式)
- (例:markers_top10_RNA_snn_res.0.1.json)
まとめ
Basepairで簡単にクラスター解析、マーカー遺伝子を同定することができます。「Single cell RNA-seq」、「Single cell RNA-seq integrate」で、1サンプルのみの解析、複数サンプルの比較も行うことができます。

参考文献
1. GitHub Pages (2024). Single-cell RNA-seq: Marker identification. GitHub Pages.