
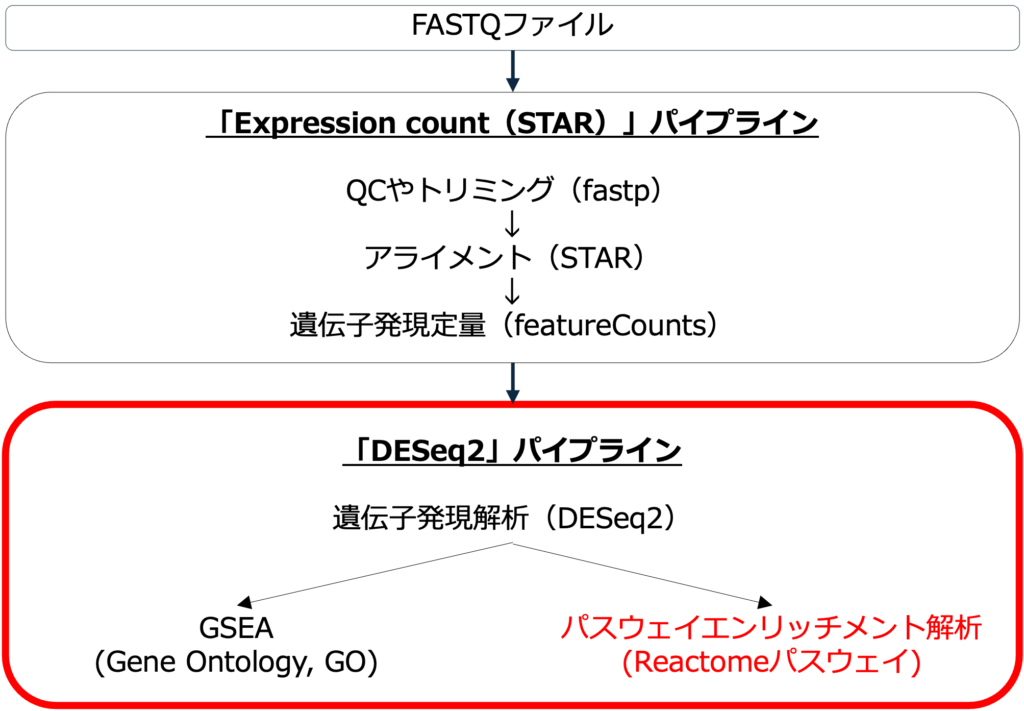
BasepairのRNA-SeqのDESeq2パイプラインは、発現変動解析、Gene Ontology(GO)エンリッチメント解析、Reactomeパスウェイエンリッチメント解析と統合されています(図1)。また、必要に応じてKEGGパスウェイエンリッチメント解析も実行できます。
DESeq2パイプラインを実行すると発現変動遺伝子のリストがアウトプットされます。このリストを用いて、DAVIDでKEGGパスウェイエンリッチメント解析を行い、KEGGパスウェイマップを可視化することができます。
DAVID(Database for Annotation, Visualization and Integrated Discovery)は無料のウェブツールです。
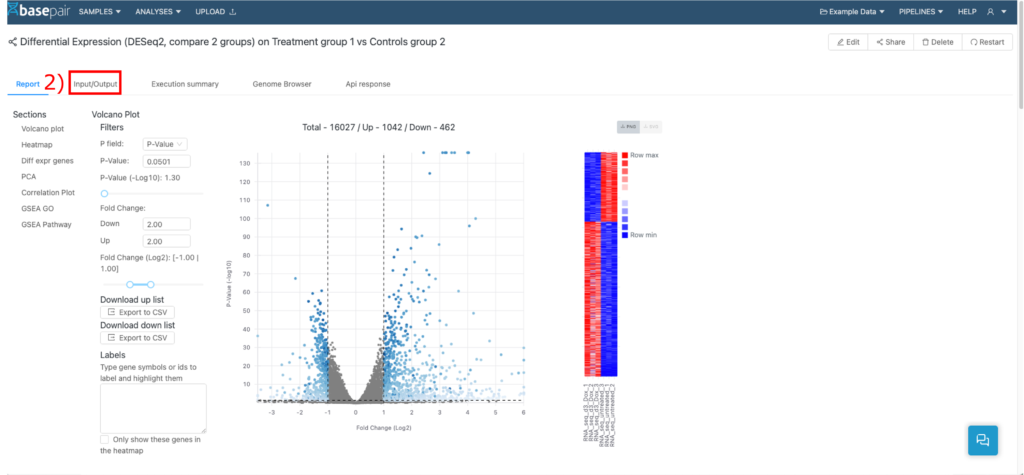
DESeq2パイプラインからのアウトプットファイルにアクセスする手順
- Basepairで解析ファイルにアクセスしてください。
- 「Input/output」タブをクリックしてください。
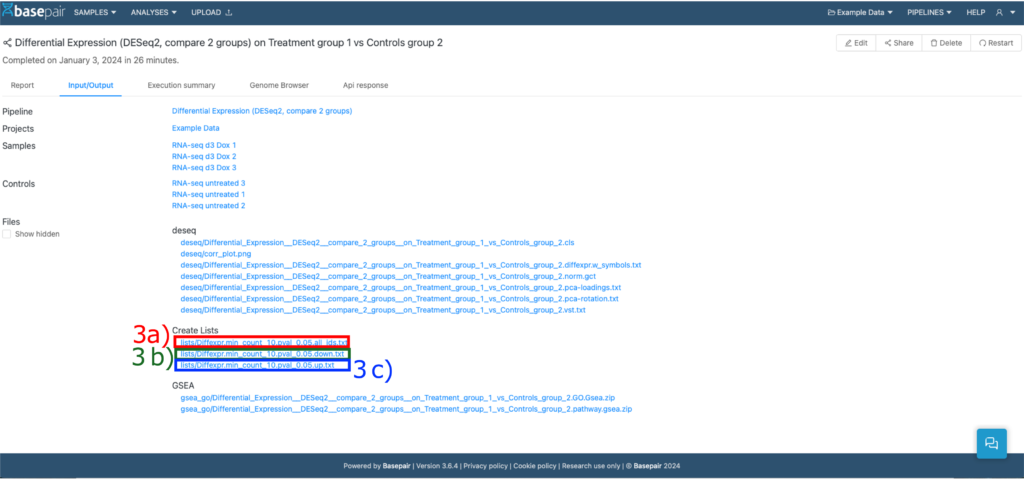
- アウトプットファイルは、「Create Lists」セクションに表示されています。ダウンロードできます。それぞれのファイルは以下の通りです。
| a) lists/Diffexpr.min_count_10.pval_0.05.all_ids.txt | 有意差のある発現変動遺伝子のリスト |
| b) lists/Diffexpr.min_count_10.pval_0.05.down.txt | DESeq2による有意なアップレギュレートされた発現変動遺伝子とその結果の表 |
| c) lists/Diffexpr.min_count_10.pval_0.05.up.txt | DESeq2による有意なダウンレギュレートされた発現変動遺伝子とその結果の表 |
アウトプットファイル「lists/Diffexpr.min_count_10.pval_0.05.all_ids.txt」は、遺伝子名が並べられたテキストファイルです。
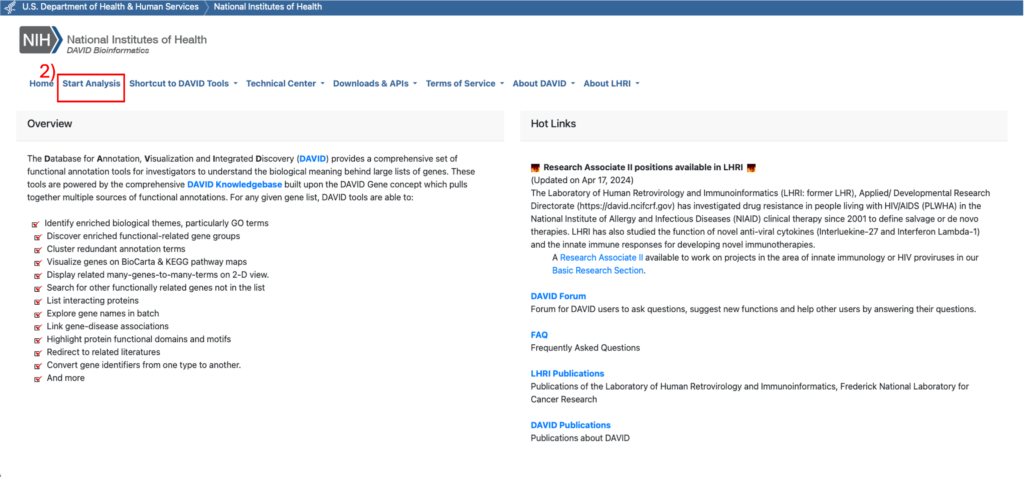
DAVIDによるKEGGパスウェイ解析の手順
- https://david.ncifcrf.gov/ にアクセス
- 「Start analysis」をクリック
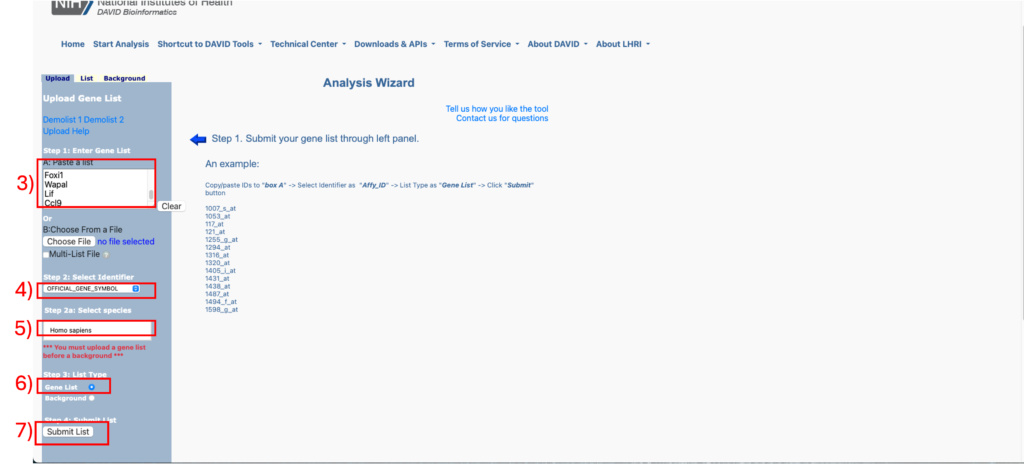
- Step 1:lists/Diffexpr.min_count_10.pval_0.05.all_ids.txtの全てをコピーして、「Paste a list」の部分にペースト
- Step 2:「OFFICIAL_GENE_SYMBOL」を選択
- Step 2a:生物種(例:Homo sapiens)を入力
- Step 3:「Gene List」をチェック
- Step 4:「Submit List」をクリック
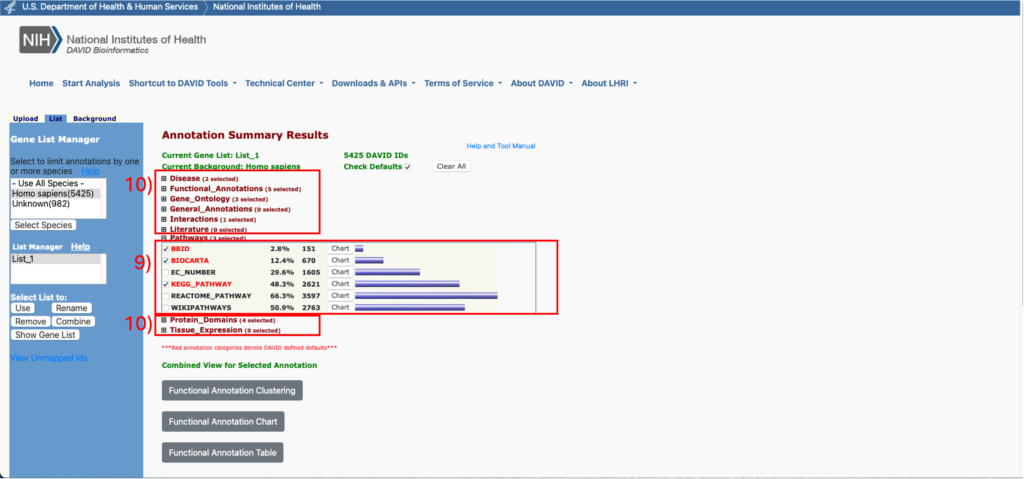
- 「Functional annotation chart」をクリック
- 「Pathways」をクリックし、「KEGG_PATHWAY」以外のオプションのチェックを外す
- その他のデフォルト選択されたオプションのチェックを外す
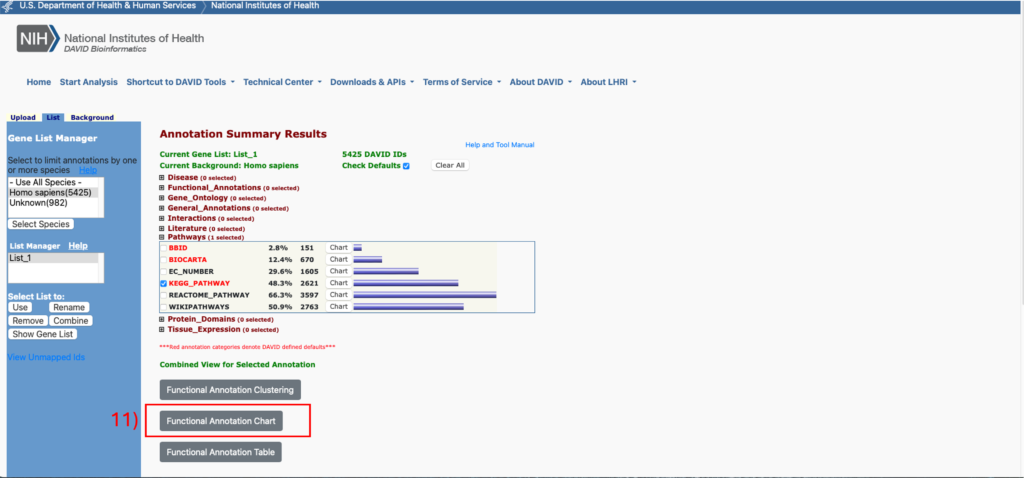
- 「Functional Annotation Chart」をクリック
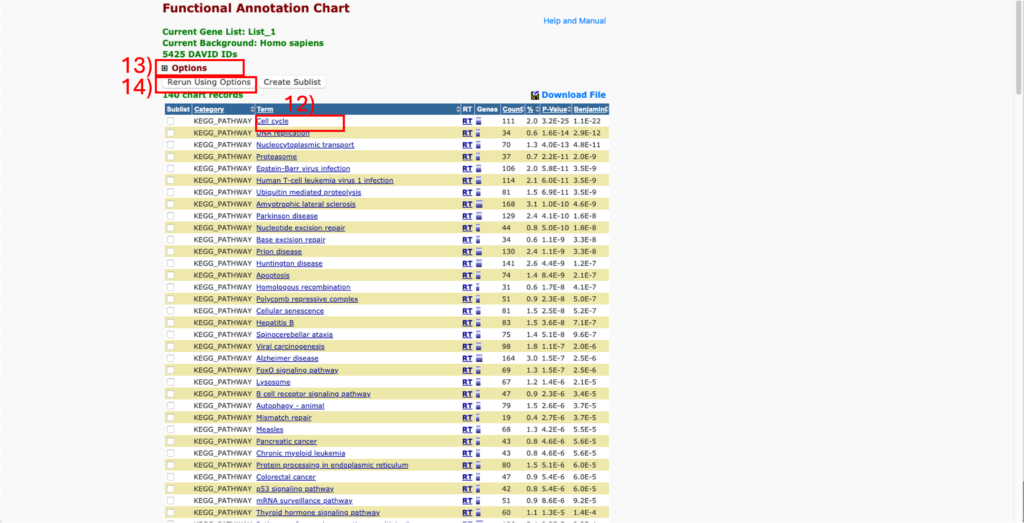
- 各パスウェイをクリックして、パスウェイに関与する遺伝子を見ることができます。
- 「Options」をクリックすると、パラメータを変更できます。
- パラメータを変更する場合は、「Rerun using options」で再実行してください。
まとめ
BasepairのDESeq2パイプラインからアウトプットされた発現変動遺伝子リストを用いて、DAVIDでKEGGパスウェイ解析を簡単に行うことができます。
最大6サンプル フリートライアル 実施中
最大6つのサンプルを無料でアップロードして分析できます。アップロードされたサンプルに対する解析は無制限です。世界トップクラスの機関、研究室、製薬チームがBasepairを使用して、数千ドルを節約している理由をご覧ください。