
NGS データ分析のボトルネック
今日の次世代シーケンス (NGS) における最大のボトルネックの1つはデータ分析です。これは、すぐに利用できる大規模なコンピューティング、数千のサンプルを並行して処理するための利用可能なインフラストラクチャー、および分析を簡素化する多数のプログラミングライブラリがあることを考えると驚くべきことです。複雑な統計演算を平易な英語に似たものに変換します。
では、なぜ私たちの多くはNGSデータの分析に数日、場合によっては数週間も行き詰ってしまうのでしょうか? Basepairの創設者でハーバード大学医学部の講師であるAmit Sinhaは、Webプラットフォームの開発に取り組み始めたとき、自身の10年間の計算生物学の経験と同僚の最も根強いハードルを振り返り、NGSデータ分析プロセスにおける不必要なボトルネックに焦点を当てました。
Amitが発見した問題は、技術リソースの不足ではなく、複数のコマンドラインソフトウェアやプログラム間のファイル変換、およびさまざまなアルゴリズムの完了を待つ無駄な時間を必要とする、バラバラなアプローチにありました。さらに、結果は数十のファイルに分散しており、興味のある情報は力ずくで掘り出さなければなりませんでした。
より高速な代替手段を実現するには、バイオインフォマティシャンが自動スクリプトを実行するためにCLIでの作業に貴重な時間を費やし、数十、数百のサンプルが関係する場合は IT 部門にワークフローの並列化を支援してもらうなど、日常的にスケジュールがあふれているチームメンバーに頼る必要がありました。これも理想的ではありませんでした。
NGSデータ分析プロセスで考えられるあらゆるボトルネックを経験したAmitは、生のfastq ファイルを取得し、選択したパイプラインに基づいて複雑な分析操作を自動的に実行するワンストップ ショップとしてBasepairを作成しました。 その結果、完成したレポートと一連のインタラクティブなグラフが作成され、研究者やバイオインフォマティシャンは、アルゴリズムの操作に行き詰まったり、スケジュールが空くのを待つことなく、下流の分析に進むことができます。
Basepairでは複数の分析が並行して実行され、すべてのコンピューティングがクラウド内の強力な仮想マシンでホストされるため、従来、複数のサンプルの分析に費やされていた数日から数週間が1時間未満に短縮されます。
Basepair Webプラットフォームの主軸は自動化されたワークフローであり、ハーバード大学医学部、スタンフォード大学、メモリアル スローン ケタリングがんセンター、その他の世界クラスの機関のチームが何千時間もの時間を節約してきました。
自動化されたワークフロー
Basepairでは、エンド ユーザーの科学者が最小限のクリックで、かつ最小限の作業時間で迅速かつ正確な結果を確実に得ることができるように、いくつかの手順を講じています。 また、NGS 研究にすぐに使用できるパイプラインとして最も有用なワークフローを自動化しており、シーケンス手法ごとにいくつかのワークフローが利用可能です。
RNA-Seq、DNA-Seq、ChIP-Seq、または ATAC-Seqデータについては、バイオインフォマティクスの最新研究をモデルにしたカスタム パイプラインを備えています。 私たち自身のバイオインフォマティシャンは、配列データが最高の精度で解釈されることを保証するために、これらのパイプラインのアルゴリズムを微調整しました。
より専門的なユーザーが懸念しているのは、仲間のバイオインフォマティシャンや研究所の所長から聞いたこんな話のような状況です。「真夜中に寝ぼけ眼で起きてNGSデータ分析パイプラインの結果を確認し、ファイルを別のファイルに変換する。フォーマットを確認し、下流分析の次のステップを準備し、気を失いながらベッドに戻る…。」 Basepairでも同じ途方もない苦労を経験したため、手動タスクを自動化できる堅牢なAPIの有用性も理解しています。これについては後ほど説明します。
ここでは、自動化されたワークフローが個々のベンチサイエンティストとあらゆる規模の研究チームの両方の作業をどのように容易にするかを理解するために、RNA-Seq で利用可能ないくつかのオプションを検討してみましょう。
RNA-Seq ワークフロー
RNA-Seq は、差次的遺伝子発現解析(Differential Expression Gene、DEG)のほか、融合イベント、遺伝的変異、新規転写物、およびその他の多数の生物学的現象の検出に最も一般的に使用されます。
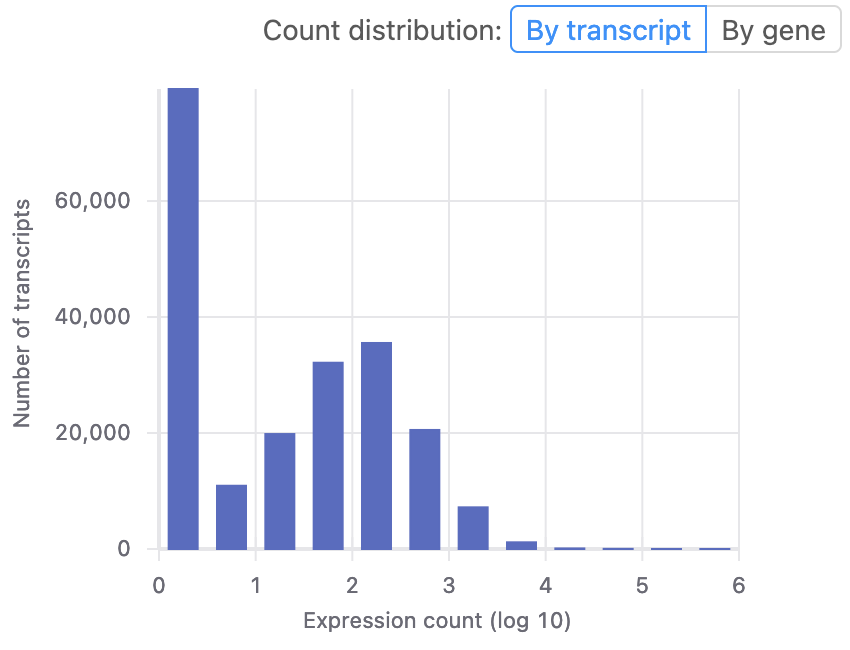
Basepairを使えば、データをアップロードした後、数回クリックするだけでアライメント、発現カウントのパイプラインを開始できます。 このパイプラインはSTARを使用して、生のシーケンス データの品質を自動的にチェックし、シーケンスの汚染物質が存在する場合はそれを除去し、データを目的のゲノムにアライメントします。生データ、あるいは正規化された遺伝子発現レベルは、featureCountsを用いて定量化され 組み込みの IGV ブラウザーでデータを簡単に表示できます。 Basepair を使用すると、サンプルが6つであっても6,000個であっても、それらを Amazon のクラウドリソースで並行して処理し、結果を迅速に提供できます。
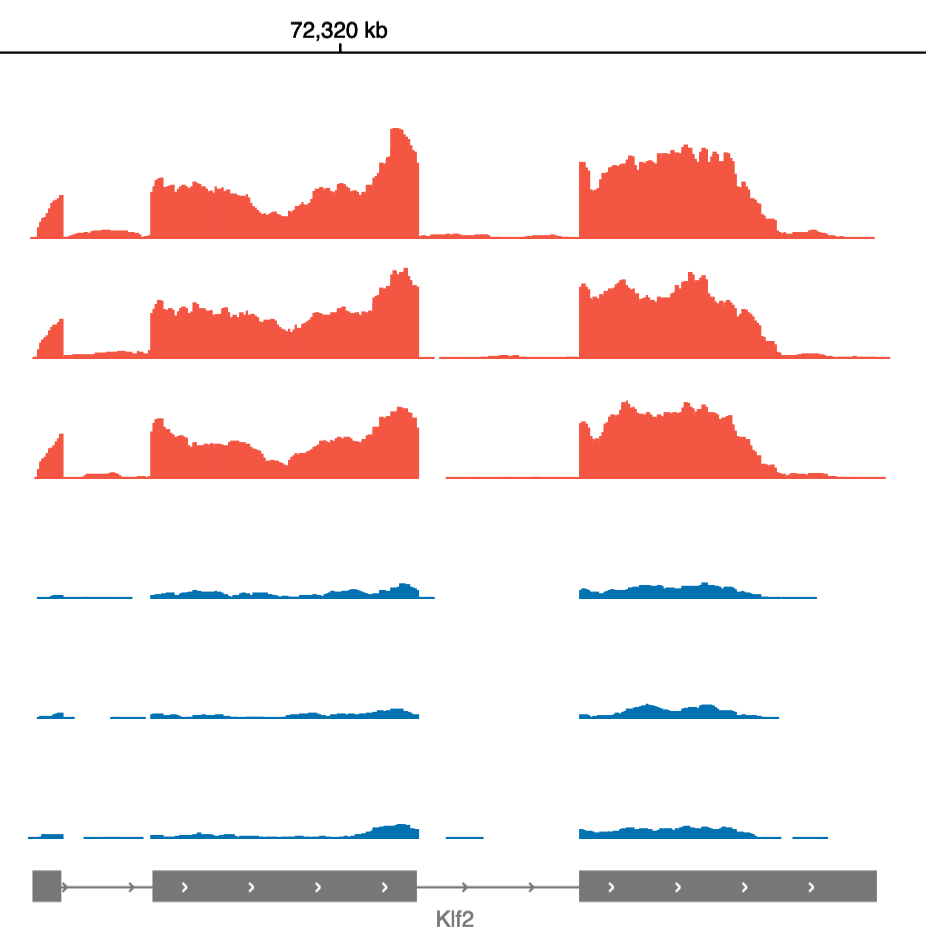

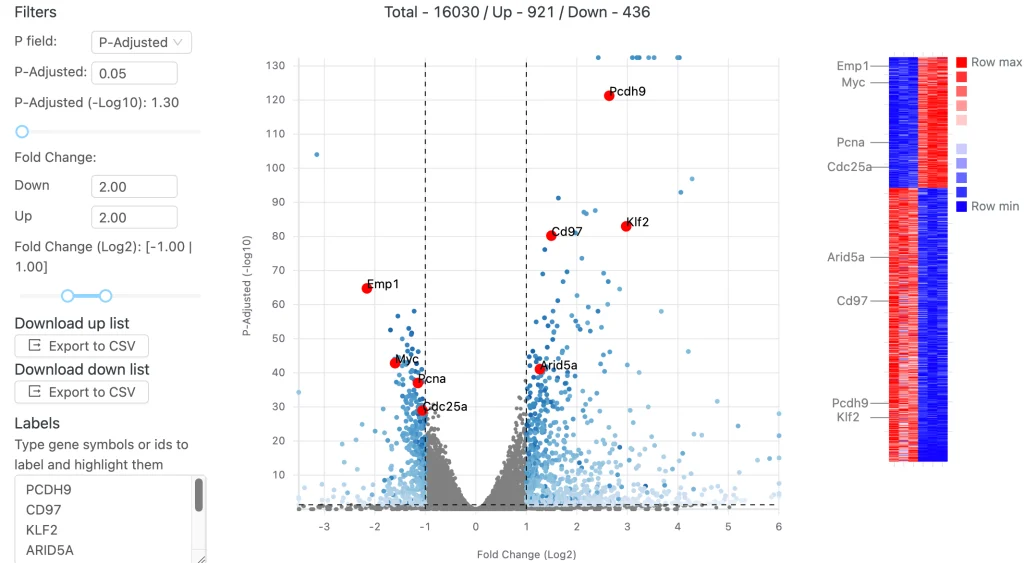
データのアライメントが完了すると、DESeq2を使用してアップレギュレートされた遺伝子とダウンレギュレートされた遺伝子を自動的に特定し、主成分分析 (PCA) を実行するだけでなく、ジーンオントロジー (GO) やGSEAを使用したパスウェイエンリッチメント解析も実行します。 これらの分析の結果は、データを迅速に閲覧できるインタラクティブプロットと表でブラウザ上に表示します。

より詳細なトランスクリプトーム解析のために、 cufflinks/cuffdiff、leafcutter 、GATK を使用したバリアント呼び出しなどのツールを使用してスプライシングを評価するパイプラインも提供しています。
API
私たちは、Pythonとコマンドライン用の強力なAPI(アプリケーションプログラミング インターフェイス)をまとめ、可能な限り読みやすく、シンプルで、直観的なものにしました。 前述したように、Basepair のグラフィカル ユーザーインターフェイスは、ワンクリックで迅速に分析できるようにすでに合理化されていますが、数十、数百、さらには数千のサンプルとなると、堅牢な API の追加に勝るものはありません。
この 追加のブログ投稿 では、Python API が大規模な NGS データ分析の自動化にどのように役立つかをいくつかの例で説明しています。 コマンドライン API を確認したい場合は、 ここに別の役立つ投稿を作成しました 。 (ヘルプ ドキュメントを表示するには、サインインする必要があります。アカウントをお持ちでない場合は、この投稿の最後にあるボタンを使用して 1 分以内に 2 週間の無料トライアルにサインアップできます。)
あなたがバイオインフォマティシャンでない場合や、開発経験がない場合でも、その投稿の例をざっと見て、Basepair の API で何ができるかを理解することをお勧めします。 Basepair は、API やあらゆる統合のセットアップもお手伝いします。詳細については、こちらからお問い合わせください。
Basepair には 50 を超えるワークフローがあり、さらに増え続けています。 私たちのチームは、実行が簡単で解釈が簡単なバイオインフォマティクス分析を提供するために、最新の研究を深く掘り下げています。 私たちは、Basepair の API をできる限り読みやすく、シンプルで、堅牢なものにしました。 優れた API は、チームが非常に短時間で数千のサンプルにスケールアップするのにも役立ちます。 強力な並列処理のおかげで、数千の分析を同時に実行できるため、追加サンプルのための時間的負債が発生しません。
当社の完全なプラットフォームを今すぐ 無料で 、 こちら にサインアップして 試し、独自の NGS データを使用して自動化されたワークフローを探索してください。
この記事は、How automated workflows simplify NGS analysisを翻訳したものです。