
エンリッチメント解析(GSEA)はRNA-Seqの際によく行われます。とはいえ、Rなどプログラミングを使用して実行するのは高いハードルがあります。また実行時間を要します。Basepairでは数回クリックするだけでGSEAを簡単に実行できます。
GSEA(Gene set enrichment analysis、エンリッチメント解析)は、事前に定義された遺伝子のセットが、コントロールと比較したときに特定のデータセット内で濃縮されているかどうかを判断するために使用される計算手法です。
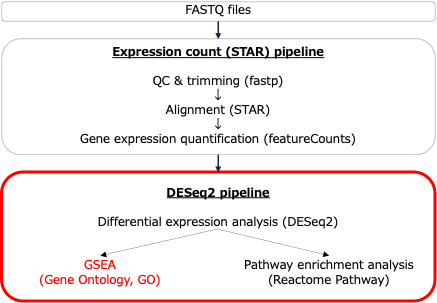
DESeq2 パイプラインでの エンリッチメント解析(GSEA)の実行
Basepairでエンリッチメント解析を行うのは非常に簡単です。GSEAはBasepairのDESeq2パイプラインにあらかじめ組み込まれています。DESeq2パイプラインは、2つ以上のサンプルグループの発現変動遺伝子を比較するために使用されます。
DESeq2の実行
BasepairでDESeq2パイプラインを実行すると、まずExpression count(STAR)パイプラインが自動的に実行されます。このパイプラインはまず、QC、データトリミング、アライメントを行ます。その際、GSEAの後のステップに必要な発現データセットファイルを作成します。次に、DESeq2パイプラインが開始し、発現変動解析が実行され、GSEAに必要なクラスファイル (cls) が生成されます。発現変動解析が終了すると、GSEAが自動的に実行されます。
「DESeq2」パイプラインを実行するには、次の3つの手順を実行するだけです。シーケンサーから出力されるFASTQ形式がインプットファイルとなります。
- FASTQファイルをBasepair にアップロード
- 「DESeq2」パイプラインを選択
- 「Run analysis」をクリック
「STAR」は「DESeq2」の前に自動的に実行されます。GSEAは「DESeq2」パイプラインで自動的に実行されます。
遺伝子セットファイルは自動抽出
GSEAに使用されるGene Ontology(GO)でアノテーションされた遺伝子セットファイル(gmt)は、Molecular Signatures Database(MsigDB)から自動的に抽出されます。したがって、GSEAを実行するためにデータを準備する必要はありません。また、両方のパイプラインで全てのパラメータが事前にセットされています。そのため、解析を実行する前に解析パラメータを設定する必要もありません。GSEAに使用されるアルゴリズムは、Broad Instituteによる説明もご参照ください。
インタラクティブなエンリッチメント解析アウトプット
DESeq2パイプラインでの解析が終了すると、インタラクティブなプロットと表を含むレポートとアウトプットファイルが生成されます。これらはすべてダウンロード可能で、論文発表やさらに下流の解析に使用できます。GSEAの結果は、エンリッチされた遺伝子セットとその統計量を含むインタラクティブな表としてレポートの中に表示されます(図2)。
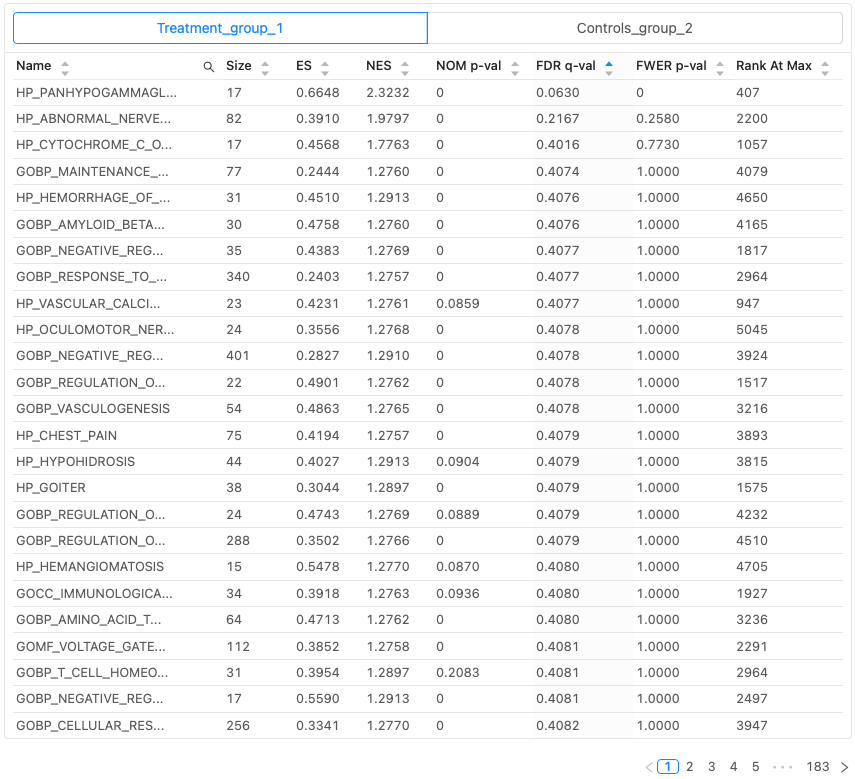
結果リストの用語
図2で使われている用語は以下の通りです:
| Name | 事前に定義された遺伝子セットの名前。 |
| Size | 定義された遺伝子セットの遺伝子数。 |
| ES | Enrichment score。ランク付けされた遺伝子リストの上位または下位で、定義された遺伝子セットがどのエンリッチしているかを反映するために使用されるスコア。 |
| NES | Normalized enrichment score。ESを遺伝子セットのサイズのばらつきで正規化したもの。 |
| NOM p-val | Nominal p-value 。あらかじめ定義された1つの遺伝子セットのESの統計的有意性。 |
| FDR q-val | False discovery rate q-value。所定のNESを持つ遺伝子セットが偽陽性である確率の推定値。 |
| FWER q-val | Family-wise error rate。結果のリストに1つの偽陽性遺伝子セットが含まれないことを保証するための統計的有意性のより保守的な推定値。 |
| Rank at max | 最大ESが発生したランクリストの位置。 |
Basepairのエンリッチメント解析の全容
結果は「Input/Output」タブを確認
すべての結果は「Input/Output」タブのGSEAの項目に格納されています。zip形式で圧縮されているので、ダウンロードして確認してください。また、これらは、論文発表や、さらに下流での結果の検証に利用できます。
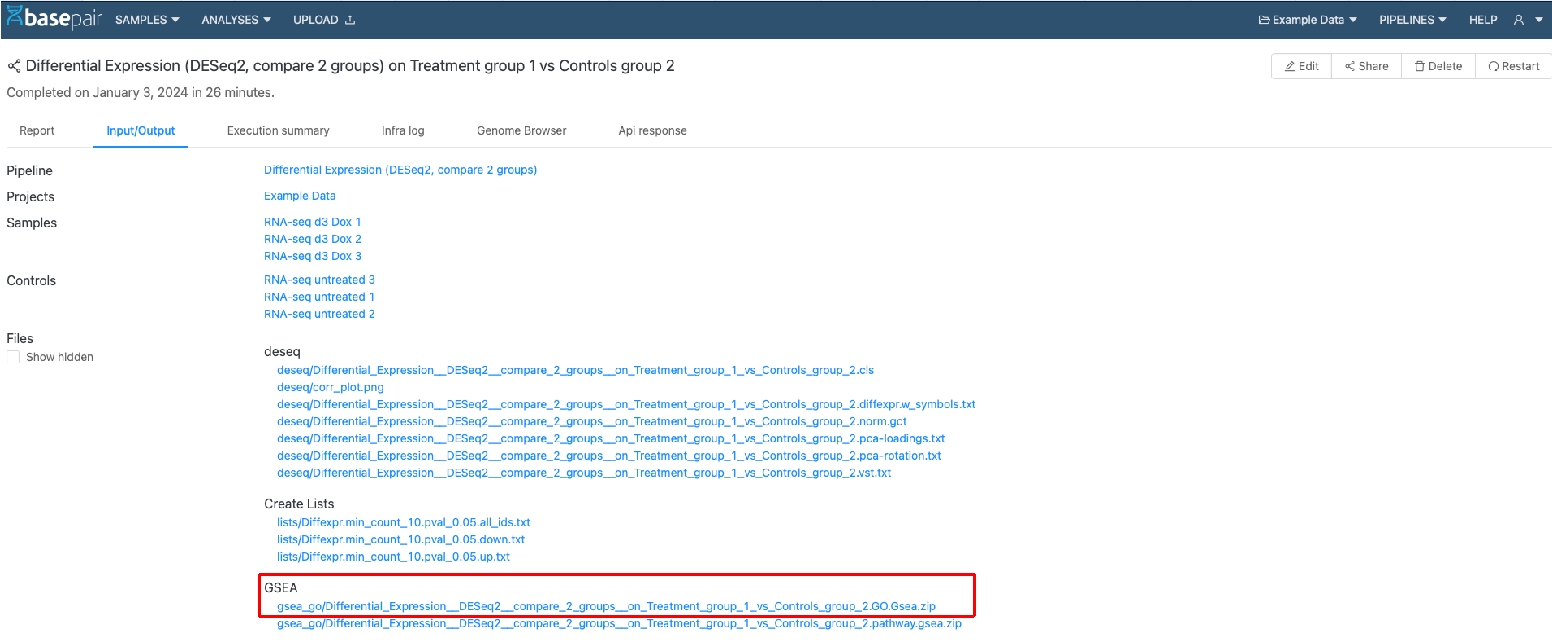
結果ファイル一覧
GSEAのzipフォルダには、以下のファイルが全て含まれています。
- フィルターされたデータセットのGSEAレポート(index.html)
- 解析されたすべての遺伝子セットの詳細なエンリッチメントの結果(html形式)
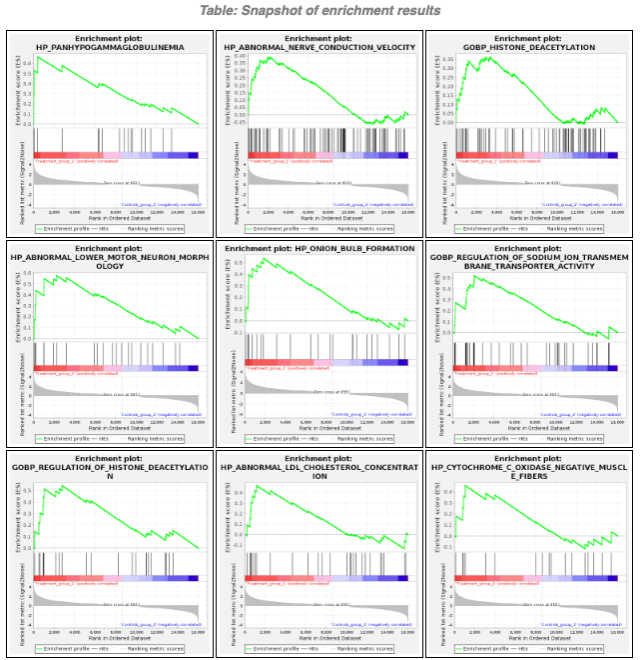
- 各遺伝子セットのエンリッチメントプロット(png形式)
- Excel形式またはタブ区切りテキスト形式の詳細なエンリッチメント結果表
- 各遺伝子セットのヒートマップ(png形式)
- 陽性エンリッチメント結果のスナップショット(pos_snapshot.html)
- 陰性エンリッチメント結果のスナップショット(neg_snapshot.html)
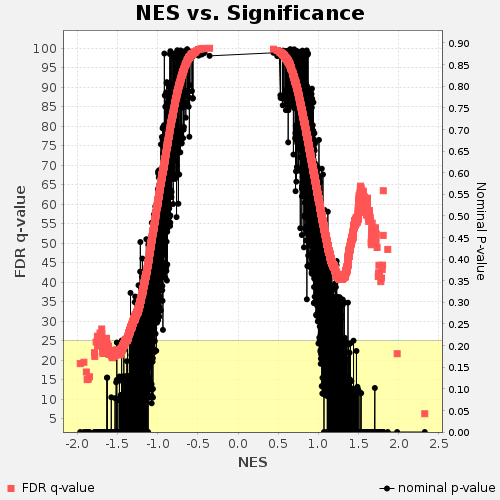
- FDRのq-valまたはNOM p-valに対するNESのプロット(pvalues_vs_nes_plot.png)
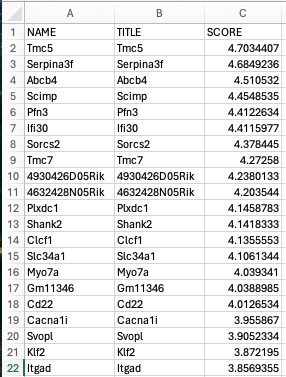
- ランク付けされた詳細な遺伝子リストとメトリックスコア(Excel形式またはタブ区切りのテキスト形式) (ranked_gene_list.tsv)
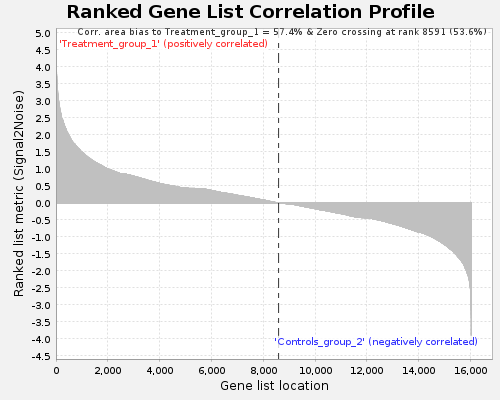
- ランク付けされた遺伝子リストのコリレーションプロット(png形式)
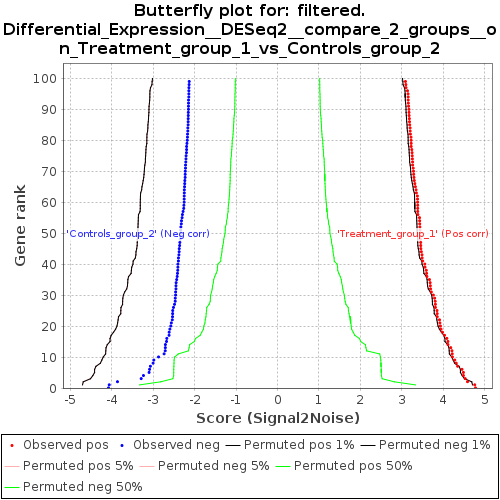
- 遺伝子ランクとランキングメトリックスコアの正負の相関を示す有意遺伝子のバタフライプロット(butterfly_plot.png)
また、「edb」という名前のサブフォルダーには以下のファイルが入っています:
- <analysis name>.cls
- filtered.rnk
- gene_sets.gmt
- results.edb
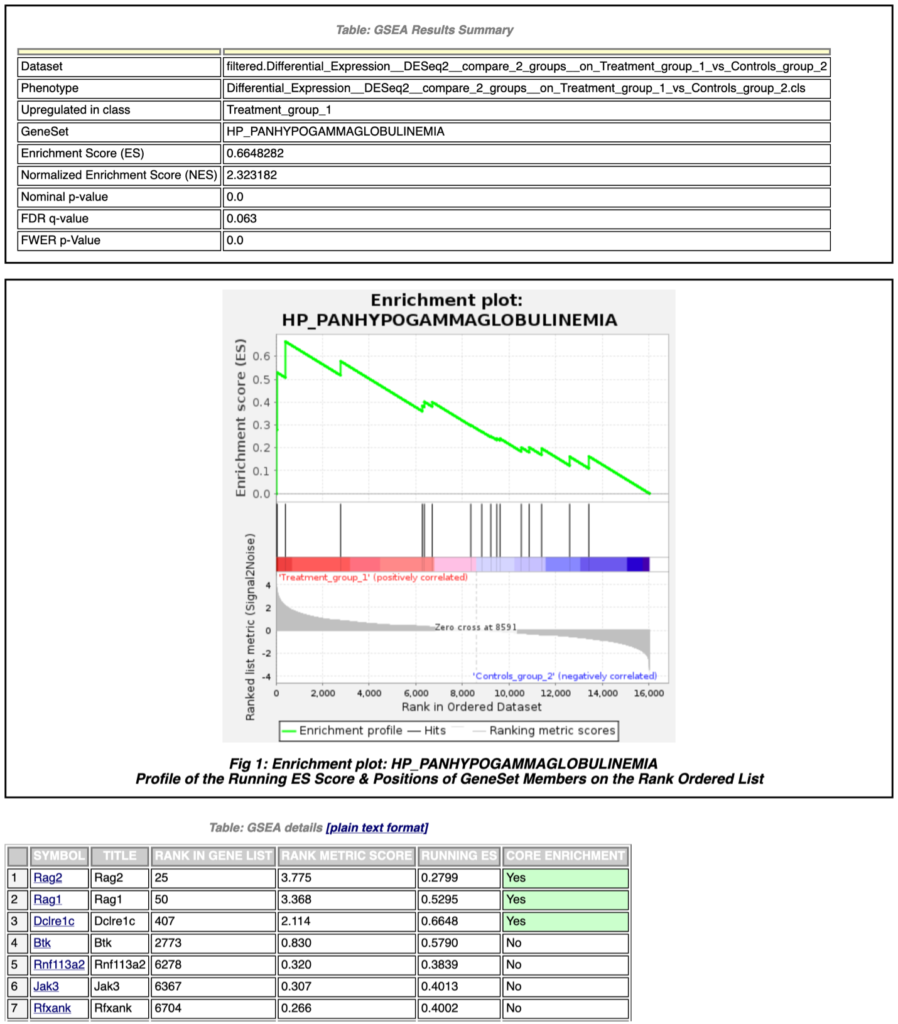
GESAレポートのスクリーンショット

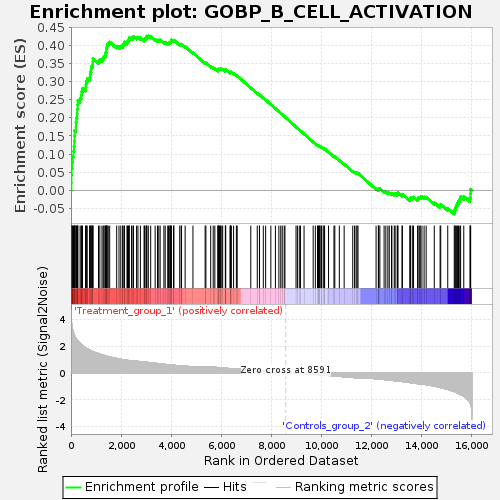
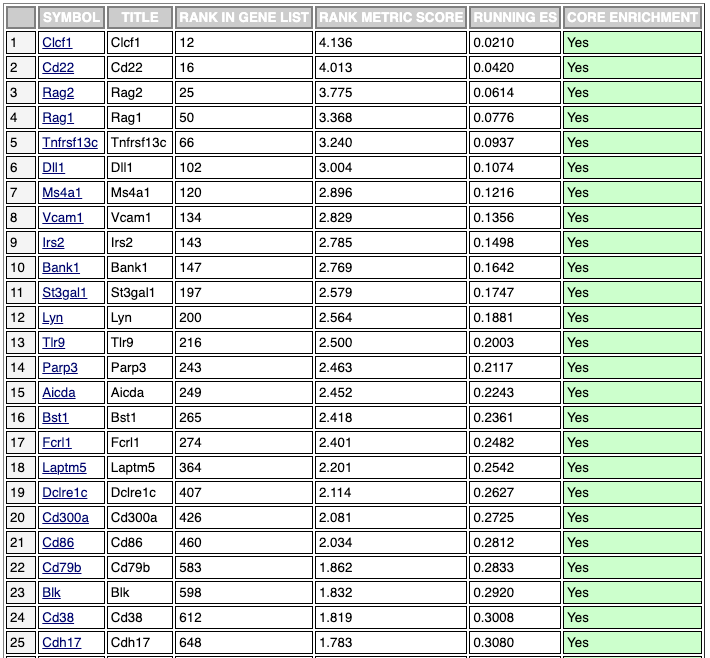
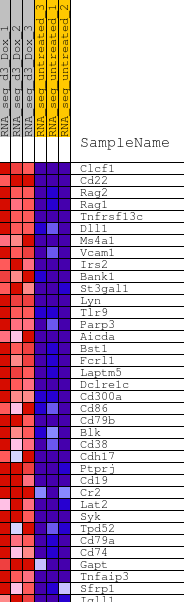
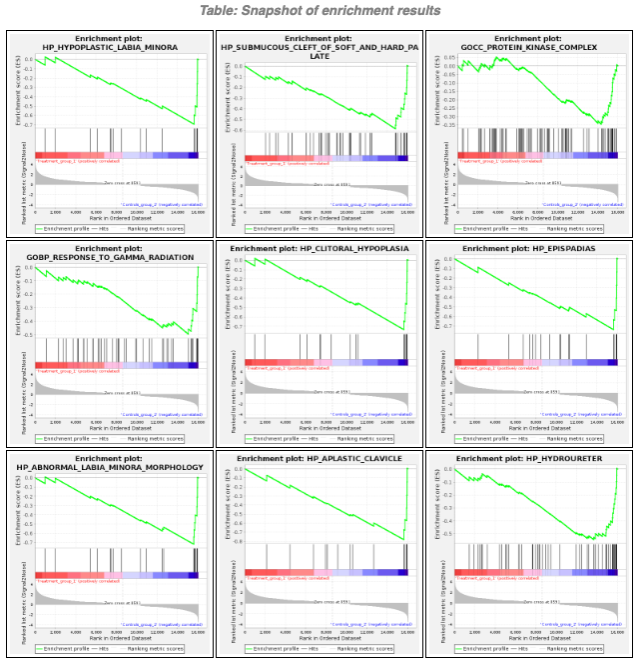
まとめ
Basepairではエンリッチメント解析を実行するのも簡単です。GSEAのためのファイルの準備とパラメータ設定の労力と時間を節約するできます。 パイプラインの実行は数回クリックするだけで、とても簡単です。
今すぐ6サンプルの無料トライアルで始めてみませんか?

参考文献
1. Broad Institute, Inc. (2004). GSEA Gene set enrichment analysis. Broad Institute, Inc.